INTRODUCTION
Exposure to chronic stress can have broad effects on health, ranging from an increased predisposition to neuropsychiatric disorders such as anxiety, depression, and dementia to the dysregulation of immune responses [1]. Compared with adulthood, adolescence is a crucial developmental stage for continued brain maturation, particularly in the limbic and cortical regions, which play a role in the physiological and emotional changes coincident with adolescence [2]. A growing number of studies indicate that stressors experienced during adolescence may affect the trajectory of neural maturation and predispose individuals to the development of mental health problems such as anxiety and depression in adulthood [2ŌĆō4]. Stress-related dysregulation of the neuroendocrine and immune systems in the development of the brain has been hypothesized as one of the major biological links between neuroinflammation and neurobehavioral dysfunction. During adolescence, exposure to psychosocial stress has been shown to upregulate inflammation by enhancing the transcription of proinflammatory genes such as interleukin-1╬▓, interleukin-6, interleukin-8, cyclooxygenase 2, and tumor necrosis factor-╬▒, as well as elicit increases in circulating levels of proinflammatory cytokines [5,6]. Through this mechanism, persistent stress may contribute to a chronic inflammatory environment, leading to negative downstream effects of adverse psychiatric events in adulthood. However, early life stress has demonstrated a clear association with psychological morbidities such as anxiety and depression in adulthood, but the impact of chronic stress during adolescence on nonpsychological trauma in adulthood remains unknown.
Traumatic brain injury (TBI) is a major global public health concern and a common cause of morbidity and mortality [7]. TBI outcomes are affected by complex and multifactorial processes, including the heterogeneous nature of the human population, different injury types and severity, and the timing and characteristics of postinjury clinical care [8].
Chronic stress and TBI have overlapping pathophysiologies, such as neuroinflammation and neurobehavioral abnormalities. Juvenile stress can affect normal neuronal maturation and increase psychological and physiological vulnerabilities downstream. We hypothesize that juvenile stress-related neuronal circuit disturbances can also influence significant neuronal injury vulnerabilities to nonpsychological neuronal stress, such as TBI. Therefore, TBI outcomes can be affected by chronic stress during adolescence.
Consequently, we investigated whether juvenile exposure to unpredictable chronic mild stress (UCMS) is associated with a more pronounced increase in neurobehavioral deficits and neuroinflammatory responses in mice exposed to nonpsychological neurotrauma with moderate-to-severe controlled cortical impact (CCI) during later adulthood.
METHODS
Ethics statements
All experimental protocols were approved by the Animal Care and Use Committee of Chungbuk National University (No. CBNUR-854-15).
Animal population
We used 8- to 12-week-old C57Bl/6 male mice in this study. Mice were housed under a standard 12-hour light-dark cycle and had ad libitum access to food and water. Mice underwent UCMS alone, UCMS followed by CCI, CCI alone, or sham operation.
Unpredictable chronic mild stress procedure
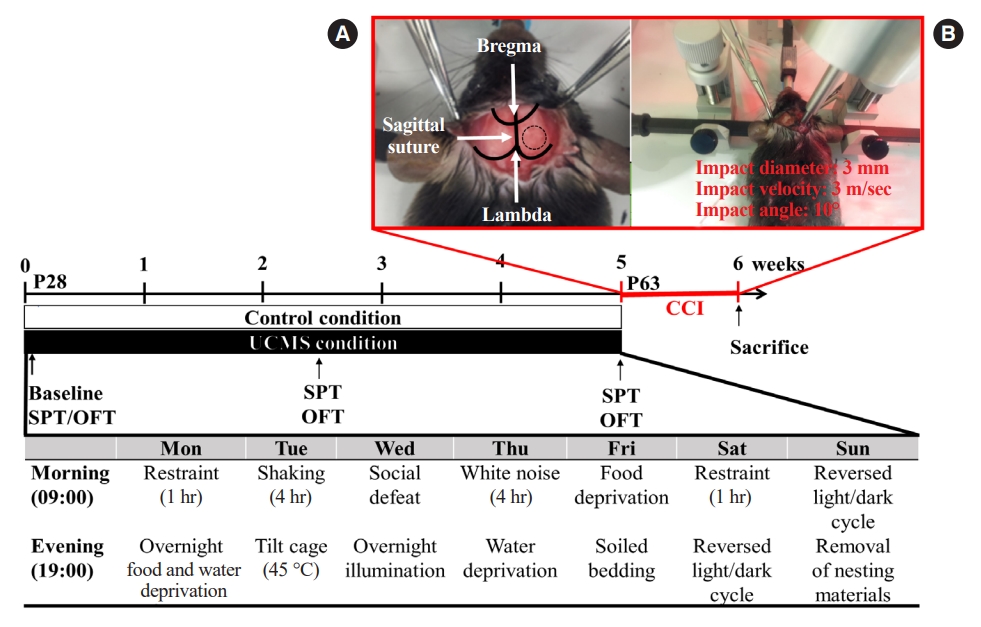
In rodents, postnatal days 21 to 59 are commonly considered ages that are associated with adolescence [4]. The UCMS procedure was performed as described previously by Jung et al. [9] with slight modifications. Postnatal day 28 mice were subjected to different stressors such as restraint, shaking, social defeat, white noise, food deprivation, inversion of the light-dark cycle, food and water deprivation, cage tilting, damp sawdust, placement in an empty cage, and overnight illumination (Fig. 1). On average, two of these stressors were applied daily at different times following a semi-random 2-week schedule. The stress procedure lasted for 5 weeks before behavioral testing. Stressors were applied during the testing phase, except on the testing days, to avoid the effects of acute stress. At least 12 hours of rest was provided between the stressor and the test.
Sucrose preference test
The sucrose preference test was performed as described previously by Zhang et al. [10] with slight modifications to evaluate anhedonia. This test was carried out before UCMS, as well as 2.5 and 5 weeks after UCMS. Mice were kept individually in separate cages and allowed to adapt to two bottles of solution (filled with 1% sucrose solution) for 24 hours. For the next 24 hours, one bottle of sucrose solution was replaced with water. The mice were then subjected to 18 hours of food and water deprivation, followed by exposure to two preweighed bottles of solution (1% sucrose solution and plain water) for 1 hour. The positions of the bottles were then changed. After the test, the weights of the sucrose solution and water consumed were recorded. Sucrose preference was calculated as follows: sucrose preference (%)=sucrose consumption/(sucrose consumption+water consumption)├Ś100%.
Controlled cortical impact
CCI was performed as described previously [11,12]. Briefly, mice were anesthetized with an intramuscular injection of 15 mg/kg tiletamine/zolazepam (Zoletil, Virbac). After cleaning the shaved head area between the ears with betadine, a midline scalp incision was made, and the right parietal bone was exposed. Next, a 4-mm-diameter circle was drawn in the center of Lambda and Bregma at 0.5 mm from the midline. Thereafter, right parietal craniotomy was carefully performed along the marked circle using a surgical microscope and micromotor drill (Stoelting). Afterward, the CCI device was calibrated relative to the exposed dura mater during craniotomy. The parameters of impact in the injured animals were a depth of 2.0 mm, a mean velocity of 3.0 m/sec, and a duration of 500 milliseconds. After the impact, the scalp incision was sutured with 5-0 nylon. The animals were then returned to their cages. The sham-operated group underwent craniotomy without CCI injury. The surgical procedure is illustrated in Fig. 1.
Behavioral testing
Behavioral testing was conducted between 8 AM and 7 PM by an observer blinded to the experimental procedures. All tests were recorded using a video tracking system equipped with Smart ver. 3.0 (Harvard Apparatus, Holliston, MA, USA), which automatically identifies postinjury behavioral changes.
Barnes maze test
The Barnes maze test was conducted as previously described with minor modifications to determine latencies and distances, errors, and speed to find the escape box [11ŌĆō13]. The maze consisted of a white acrylic circular platform (diameter, 91 cm) with 20 equally spaced holes and a black acrylic escape box (20├Ś5├Ś6 cm) along the perimeter. The maze was surrounded by four spatial cues at its height.
Acquisition trials
Each mouse was trained by way of four acquisition trials per day over 3 days with an intertrial interval of 10 to 15 minutes. Immediately before the first trial, a mouse was placed in the middle of the maze in a black starting cylinder (diameter, 10 cm), and a buzzer (80ŌĆō90 dB) was turned on. After 10 seconds, the chamber was lifted, and the mouse was pretrained to enter the escape box by guiding it to the escape box and allowing it to remain there for 2 minutes. The first trial was initiated following the pretraining trial.
At the beginning of each trial, a mouse was placed in the same starting chamber, and 10 seconds after turning on the buzzer and light, the chamber was lifted, and the mouse was free to explore the maze. The trial ended when the mouse entered the goal tunnel or 3 minutes into the trial. Immediately after the mouse entered the tunnel, the buzzer was turned off and the mouse was allowed to stay in the tunnel for 1 minute.
After each trial, the entire maze was cleaned with 70% alcohol and rotated to eliminate intramaze cues. The trials were recorded using a video tracking system equipped with Smart ver. 3.0.
Probe trial
During the probe trial, the escape tunnel leading to the target box was closed. The 90-second probe trial was conducted on day 6, and the mice were allowed to explore the maze and visit the target hole. The latency and distance to reach the target hole were recorded for the first time.
Open field test
Locomotor activity was measured in a white open-top acrylic box (40├Ś40├Ś40 cm) with an illumination intensity of 20 lux at the box floor level for 30 minutes. The activity was automatically recorded using a video tracking system equipped with Smart ver. 3.0. Distance moved (mm), time spent in the center (25%), time spent in the area margins, and mean walking speed (cm/sec) were also evaluated.
Light-dark transition test
The apparatus consisted of black (20├Ś40├Ś40 cm) and white compartments (20├Ś40├Ś40 cm) separated by a connecting gate (5├Ś8 cm). Each animal was individually placed at the center of the bright compartment (facing away from the door), and the following parameters were measured for 5 minutes: latency of the initial movement from the light to the dark area (latency of transition), the total number of transitions between the light and dark areas, and total time spent in the light area.
Western blotting
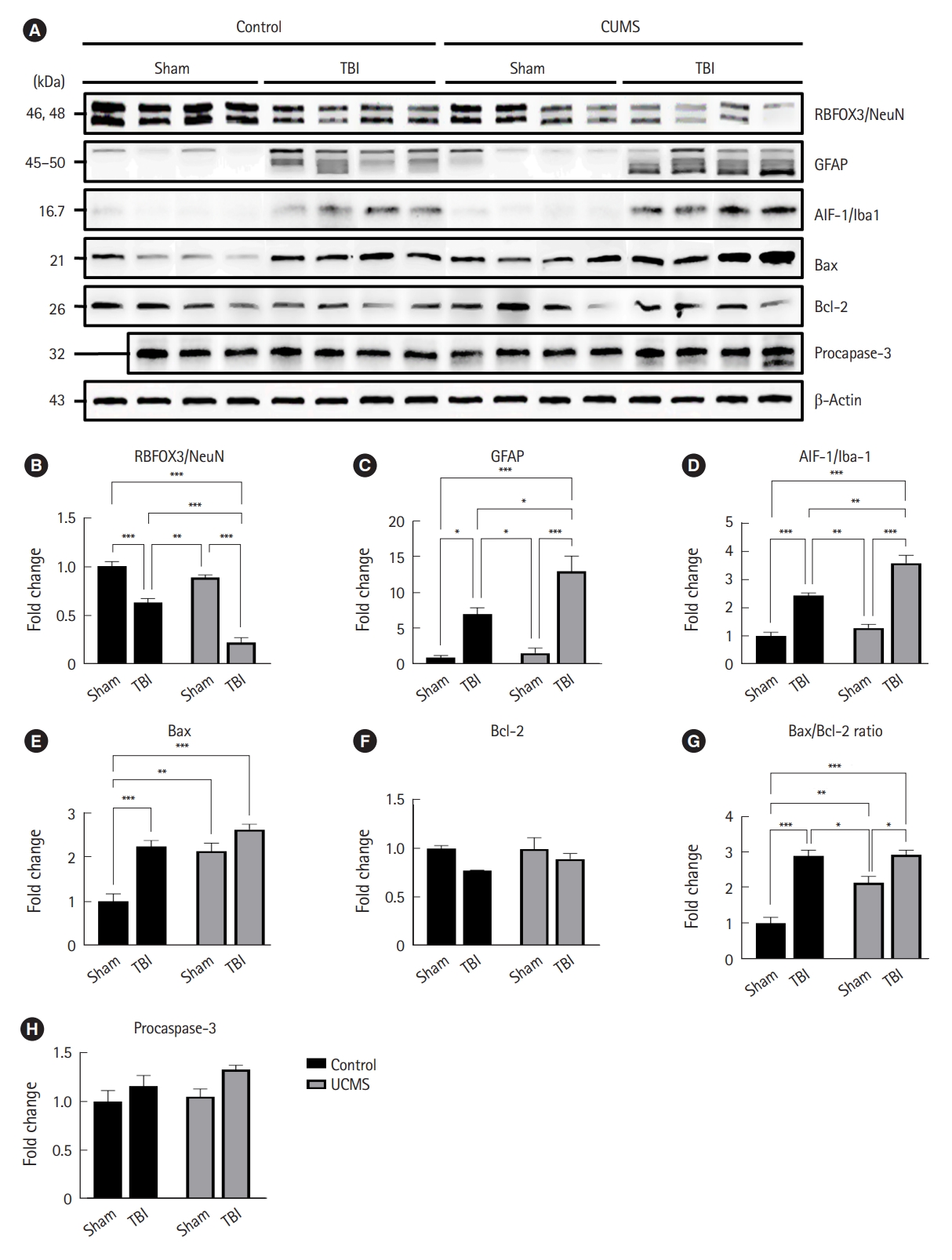
Seven days postinjury, the animals were sedated with an intramuscular injection of 15 mg/kg tiletamine/zolazepam (Zoletil) and sacrificed. Brain tissue (three to four in each group) was dissected and stored at ŌĆō80 ┬░C immediately before use. The samples were lysed with ice-cold radioimmunoprecipitation assay buffer (#MB-030-0050, Rockland) supplemented with 10 ╬╝L/mL protease and phosphatase duo inhibitor cocktail (#P3300-001, GenDEPOT) and homogenized using a syringe with a 23G needle (approximately 10 to 20 times) at room temperature (RT). The tissue lysates were incubated on ice for 30 minutes and mixed using a vortex device every 5 minutes. The supernatants were collected after centrifugation at 13,000 rpm at 4 ┬░C for 10 minutes. The Bradford assay method (#5000202, Bio-Rad Laboratories) was used to measure the protein concentration of the samples. After protein quantification, each sample was treated with 4├Ś Laemmli sample buffer (#161-0747, Bio-Rad Laboratories) containing 10% (volume/volume) ╬▓-mercaptoethanol (#M3148, Sigma-Aldrich). After boiling in a dry bath at 95 ┬░C for 10 minutes, the samples were stabilized on ice for 5 minutes and centrifuged at 13,000 rpm and 4 ┬░C for 10 minutes. Next, the supernatants (10 ╬╝g in a 4-, 7.8-, 10-, or 13.4-╬╝L volume) were separated using Any kD Mini-PROTEAN TGX Precast Protein Gels (#456-9034, Bio-Rad Laboratories) and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (#165-8004, Bio-Rad Laboratories) at a constant voltage of 200 V for 35 minutes, and then transferred to a 0.45-╬╝m (#10600023, Amersham) or 0.2-╬╝m (#10600021, Amersham) pore size hydrophobic bond polyvinylidene fluoride transfer membrane using a wet-tank (#TE22, Hoefer Inc) transfer method at 250 mA for 60 minutes. Thereafter, the membranes were incubated with 15 mL of 5% (weight/volume, w/v) bovine serum albumin (BSA; #A0100-010, GenDEPOT) in Tris-buffered saline with Tween 20 (TBS-T) and 0.1% Tween-20 (#274348, Sigma-Aldrich) in 1├Ś TBS buffer (#TR2008-100-00, Biosesang) to block nonspecific reactions for 2 hours at RT using a laboratory shaker (20 rpm; #AD-ST, GYROZEN) and washed three times with 15 mL of TBS-T for 10 minutes at RT (40 rpm). After washing, the membranes were incubated overnight at 4 ┬░C with 10 mL of 5% (w/v) BSA in TBS-T containing an appropriate concentration of the primary antibodies against RNA-binding protein, including fox-1 homology 3/neuron-specific nuclear protein (RBFOX3/NeuN), glial fibrillary acidic protein (GFAP), allograft inflammatory factor 1/ionized calcium-binding adapter molecule 1 (AIF-1/Iba-1), B-cell lymphoma 2 (Bcl-2)-associated X protein (Bax), Bcl-2, procaspase-3, and ╬▓-actin for 18 to 24 hours, and then three times rinsed with 15 mL of TBS-T for 10 minutes at RT. After washing, the membranes were incubated with 15 mL of 5% (w/v) BSA in TBS-T, including horseradish peroxidase-conjugated secondary antibodies: goat anti-rabbit (1:3,000; #170-6515, Bio-Rad Laboratories) or goat anti-mouse (1:3,000; #170-6516, Bio-Rad Laboratories) polyclonal antibodies, for 1 hour at RT (20 rpm), and then washed three times with 15 mL of TBS-T for 10 minutes at RT. After washing, the bands were visualized using Clarity Western enhanced chemiluminescence Substrate (#170-5060, Bio-Rad Laboratories) and a ChemiDoc XRS+ Imaging System (#170-8265, Bio-Rad Laboratories) according to the manufacturerŌĆÖs protocol. Then, protein bands were analyzed quantitatively using ImageJ ver. 1.53k (US National Institutes of Health), and the results were used for further statistical analysis. The expression levels of the target protein in the cortex of mice were determined relative to ╬▓-actin as an internal loading control, and all relative band intensities of target protein were normalized to the mean relative band intensity of the control and sham groups.
Statistical analysis
All data are presented as the mean┬▒standard error of the mean and, to ensure data accuracy, all western blot studies were repeated three to six times. GraphPad Prism ver. 9.3.1 (GraphPad Software) was used to analyze the normalized data and construct histograms. After using the Shapiro-Wilk and Brown-Forsythe tests to confirm data normality and homogeneity of variances, respectively, a two-way (stress, control vs. UCMS; UCMS├Śoperation, sham vs. TBI) analysis of variance (ANOVA) was used to analyze the effects of stress and brain damage. After the ANOVA verified the interaction effect (P<0.05) between stress and the operation, TukeyŌĆÖs (equal sample size) or BonferroniŌĆÖs (unequal sample size) post hoc multiple comparison test was used to analyze the differences between groups. In the post hoc test for the western blot analysis, a value of P<0.05 was considered statistically significant.
RESULTS
UCMS reduced body weight gain, sucrose preferences, and locomotor activity
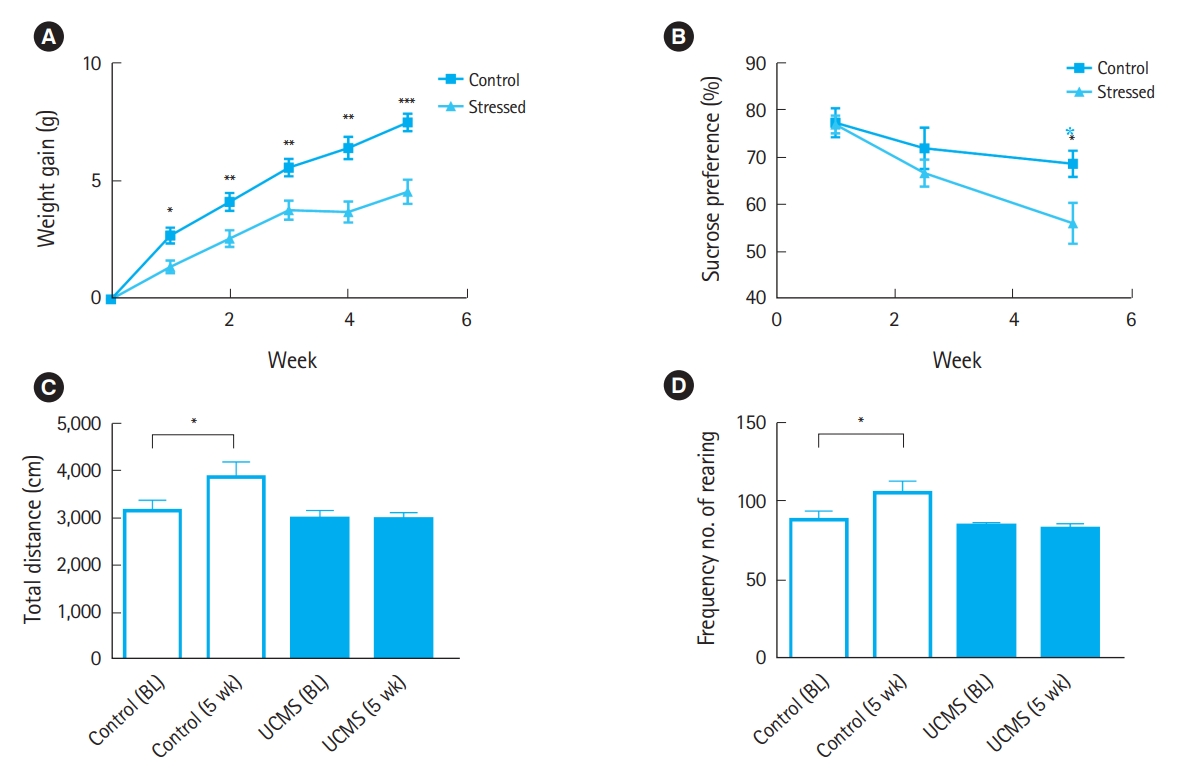
As illustrated in Fig. 2, body weight gain, sucrose preference, and locomotor activity were monitored to control for stressor efficacy. Before stress exposure, body weight gain did not differ significantly between the control and stress groups. However, stressed animals exhibited a reduction in body weight gain compared with controls after 1 week of stress exposure (0.8┬▒0.4 g vs. 2.7┬▒1.2 g, P<0.05). After 5 weeks of UCMS, body weight gain in the stress group was significantly lower than that in the control group (3.0┬▒1.5 g vs. 6.9┬▒2.8 g, P<0.001). Sucrose preference was similar between the two groups before stress exposure and after 2.5 weeks of UCMS. However, after 5 weeks of UCMS, stressed animals exhibited a significant decrease in sucrose preference (56.1%┬▒10.5% vs. 68.1%┬▒18.4%, P<0.05). In the open field test (OFT), total ambulation and rearing time significantly increased (P<0.05) in control mice after 5 weeks, whereas stressed mice did not show a significant increase in total ambulation and rearing time.
UCMS in adolescence alters neurobehavioral responses following TBI in adulthood
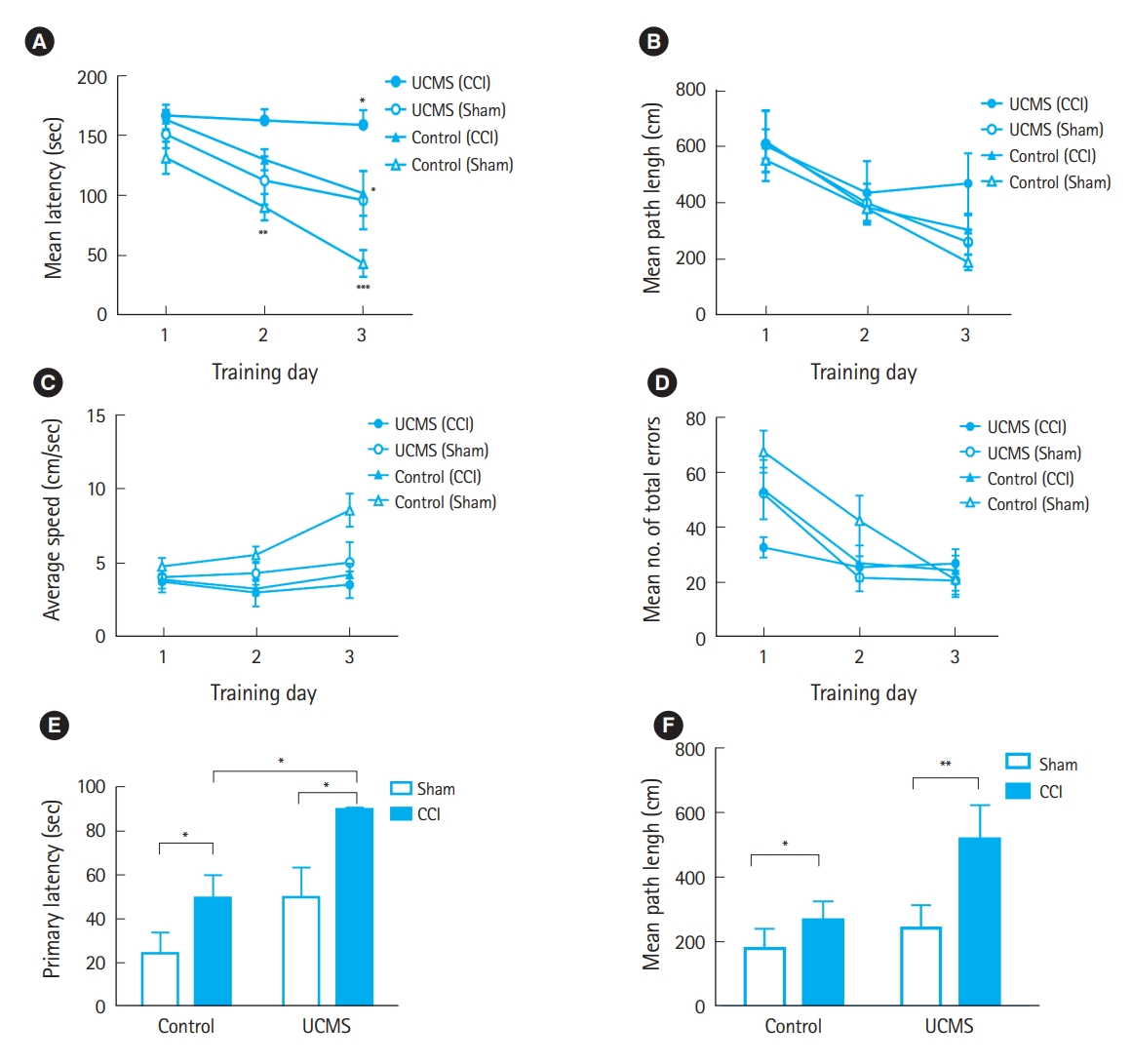
To analyze the influence of juvenile stress on spatial learning and memory deficits following TBI, we performed a Barnes maze test. During the training days, the latency (sec) to enter the target hole (total latency) was measured and analyzed using a two-way ANOVA. Nonstressed sham rats showed a significant decrease in latency during the 3 training days, indicating that they learned the task over the 3-day training period (Fig. 3A). However, mice that were subjected to TBI spent more time in the arena than those in the other groups (Fig. 3A). There was no significant difference in mean path length, average speed, and mean total errors among groups in the acquisition trials (Fig. 3BŌĆōD). The mean path length (cm) on day 5 of the retention trials in the retention phase, the mean latency, and the path length to the target stress during the Barnes maze performance were affected by TBI and/or chronic stress. Unpaired t-test demonstrated statistically significant differences in primary latency to the target hole and adjacent holes between nonstressed mice following TBI and stressed mice following TBI, indicating that stressed mice following TBI had significant retention memory deficits (Fig. 3E). In addition, the primary path length to the target hole was significantly greater in the difference between groups in stressed mice than in the difference between groups in unstressed mice (Fig. 3F).
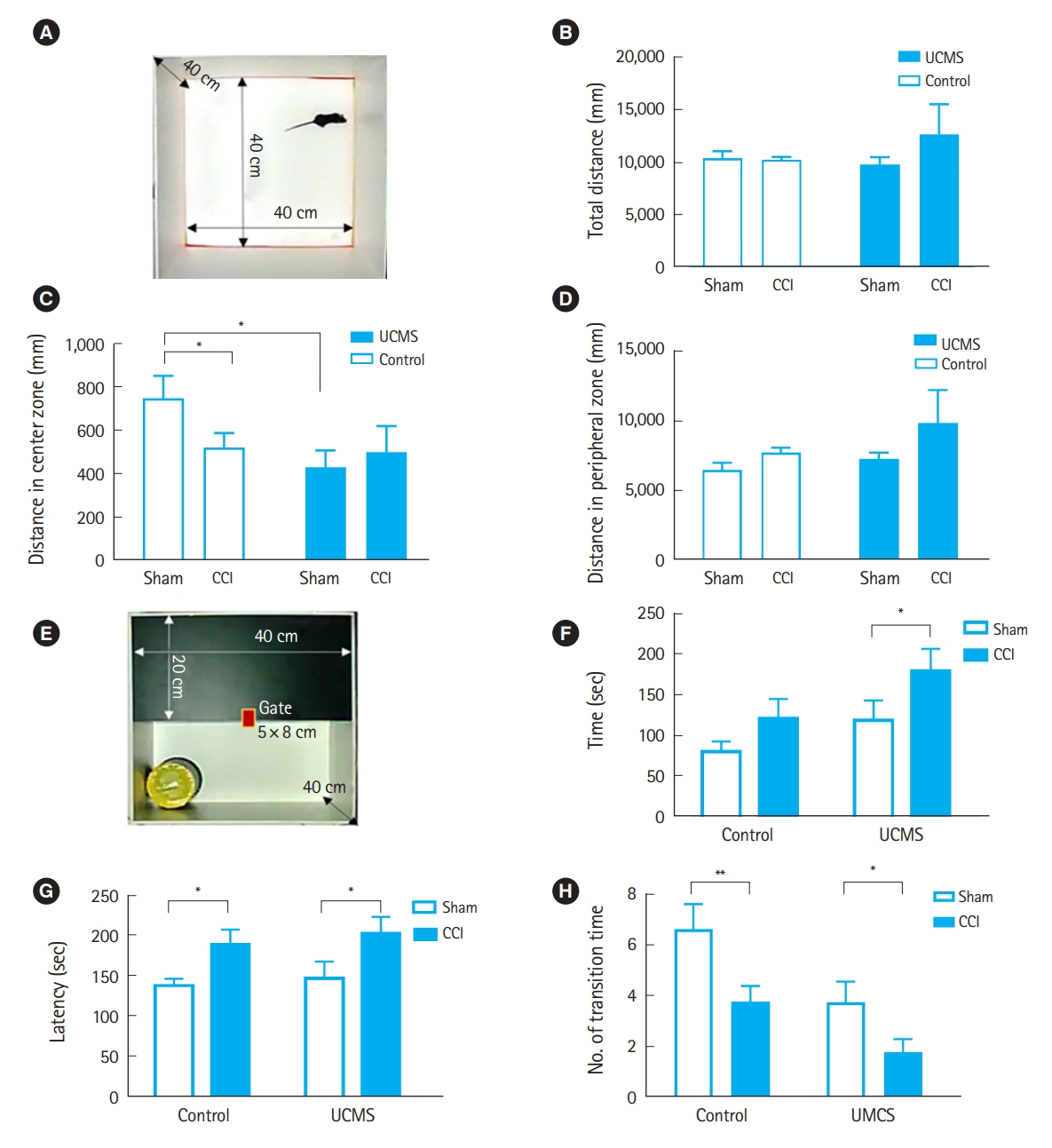
To evaluate the influence of juvenile stress on anxiety-like behavior following CCI in adulthood, we performed the OFT and light-dark transition test (LDT). Fig. 4A depicts the OFT. As shown in Fig. 4B, the total locomotor activity was not influenced by TBI or chronic stress; however, the time spent in the central region of the testing chamber between nonstressed mice exposed to TBI and stressed mice was significantly reduced compared with that for nonstressed sham mice, indicating anxiety-like behavior, although the time spent in the peripheral region of the testing chamber among different groups was not significant (Fig. 4C, D). Fig. 4E shows a schematic representation of the LDT. After 2-mm CCI, stressed mice spent significantly more time in the light compartment than nonstressed mice (Fig. 4F). Mice exposed to TBI showed significant differences in the initial latency of transition (Fig. 4G) and transition time (Fig. 4H) compared with sham mice.
UCMS in adolescence can potentiate neuronal injury and glial reactivity following TBI in adulthood
Although neuronal injury and glial reactivity did not differ significantly between the nonstressed and stressed sham groups, western blot analysis revealed that mice exposed to TBI had significantly different changes in neuronal injury and glial reactivity depending on whether they were stressed in adolescence.
Neurons are highly sensitive to traumatic injury. Neuronal injury and loss were evaluated by immunoblotting for NeuN, a neuronal-specific nuclear protein. Fig. 5A shows representative photomicrographs of NeuN western blotting in the injured cortex of each animal group. TBI caused a significant decrease in NeuN immunoreactivity compared with that in nonstressed and stress sham mice (Fig. 5B). Importantly, following TBI, the relative band intensity of NeuN in stressed mice decreased significantly more than that in nonstressed mice (P<0.001) (Fig. 5B).
To assess the effect of juvenile stress on astrogliosis following TBI, we immunoblotted GFAP. Fig. 5A shows representative images of GFAP western blotting in the injured cortex of each animal group. We found a significantly higher band intensity in the injured animals than that in both the nonstressed and stressed sham mice (Fig. 5C). Importantly, in the injured cortex, stressed mice had higher levels of GFAP than those in nonstressed mice after TBI (P<0.05).
To assess the contribution of microgliosis to TBI following juvenile stress, we performed immunoblotting for Iba-1, a macrophage/microglia-specific calcium-binding protein. Fig. 5A shows representative photomicrographs of Iba-1 western blotting in the injured cortex of each animal group. Sham animals had very low levels of Iba-1 staining, with a marked increase in all groups following TBI (Fig. 5D). Stressed mice exposed to TBI exhibited a significant increase in Iba-1 staining compared with nonstressed mice, although there were no group differences between nonstressed and stressed sham mice.
Taken together, these findings suggest that in addition to primary neuronal loss due to TBI, secondary neuronal loss caused by chronic stress may occur in the penumbra connected to the damaged region through altered biochemical conditions, such as astrocyte and microglial overactivation, indicating that stressed mice are more vulnerable to secondary neuronal loss.
UCMS in adolescence can enhance apoptosis following CCI
The levels of Bax and the Bax/Bcl-2 ratio, which are linked to biochemical circumstances in humans, also corroborate this statement. An increase in the Bax/Bcl-2 ratio indicates the probability of apoptosis in a tissue or specific region because Bax is a proapoptotic marker and Bcl-2 is an antiapoptotic marker. Levels of Bax were found to be considerably higher in either the stress-exposed or TBI-induced mice groups, or both (Fig. 5E). Additionally, the Bax/Bcl-2 ratio was significantly higher in both the stress-exposed and TBI-induced mice groups, or both (Fig. 5G). These findings suggest that apoptosis occurs under UCMS conditions and that this biochemical change with astrocyte and microglial activation may result in neuronal loss. Caspase-3 is an apoptosis executioner protein, and cleaved caspase-3 was used to evaluate caspase-3 activation as an essential apoptosis marker. However, using western blot analysis, we were unable to detect cleaved caspase-3 levels in these experiments (no data are shown in this study), and there was no interaction or significant difference in Bcl-2 and pro-caspase-3 levels between the groups (Fig. 5F, H).
DISCUSSION
Our study shows for the first time that chronic stress in the developing brain exacerbates neurobehavioral dysfunction and neuroinflammatory responses in mice exposed to moderate-to-severe TBI that occurs in adulthood. We observed that chronically stressed juvenile mice showed a greater decrease in spatial learning and memory following TBI than nonstressed mice, indicating that chronic stress influences injury progression following TBI. Furthermore, our study identified that chronic juvenile stress could potentiate glial reactivity, neuronal injury, and apoptosis following moderate-to-severe TBI that occurs in adulthood, suggesting a prime factor influencing neuroinflammation following TBI in later adulthood.
Adolescence is a time of continued brain maturation, particularly in the limbic and cortical regions, which undoubtedly play a role in the physiological and emotional changes that coincide with adolescence [2]. Exposure to chronic stress in the developing brain can be particularly harmful and lead to more brain alterations and physiological disruptions that impact health and developmental outcomes throughout life than in adults because of vulnerability to the effects of chronic stress [14].
The UCMS rat model is a renowned rodent paradigm used to induce depressive-like and anxiety-like behaviors and consists of random, intermittent, and unpredictable exposure of animals to various stressful situations, usually for at least 4 weeks [1,15]. UCMS application during mouse adolescence induces long-term depressive-like susceptibility through impairment of the equilibrium of the hypothalamic-pituitary-adrenal axis and subsequent enhanced sympathetic activation, unbalanced reactivity, and hypercortisolemia, similar to the pathophysiology in humans [1,16,17]. UCMS is a potentially reliable model to explore the association of depressive-like behavior in mice with changes in peripheral proinflammatory cytokines, as well as neuroinflammation in various regions of the mouse brain known to be involved in the pathophysiology of depression [18].
In experimental animal models, juvenile stressed mice showed behavioral abnormalities such as increased anxiety-like behavior, decreased spatial memory, increased corticosterone secretion, and altered hippocampal size after maturation, resembling those seen in neuropsychiatric disorders and increased brain vulnerability [19]. We also observed that chronic mild stress in C57BL/6 juvenile mice induces a chronic stress response, as revealed by abrogated body weight gain, decreased sucrose preferences, and stress-associated behavioral alterations, including the potentiation of anxiety and depression-like behaviors and a reduction of exploratory behavior, as well as subtle stress-related changes in spatial learning and memory function in adulthood. The UCMS used in our study, first introduced by Katz et al. [20] and subsequently developed by Willner [16], is a renowned rodent paradigm applied to mice and rats in myriad studies to induce behavioral deficits such as anhedonia and behavioral despair [15,21]. During the procedure, adolescent animals were chronically exposed to various unpredictable mild stressors. One of the more prominent tests conducted following UCMS is the sucrose preference test, which is based on the rodentsŌĆÖ innate preference for sweetened solutions rather than water and is widely acknowledged as an essential translational model for assessing anhedonia [15]. Other notable outcome measures that are highly incorporated in the UCMS literature are the OFT (measuring exploratory and anxiety-like behaviors and locomotor activity) and the LDT (measuring anxiety-like behavior) as behavioral tests also applied in the present study [16].
Severe TBI presents with significant cognitive and/or affective dysfunction, which can have perpetual adverse consequences on the quality of life. Cognitive problems following TBI include decreased memory and learning abilities, impaired attention and concentration, reduced processing speed, word-finding difficulties, and impaired executive functioning [22]. Although the cognitive processes that result in memory impairment following TBI are not fully understood, alterations in neural circuits associated with memory function contribute to memory impairments caused by TBI [23]. We previously reported that a 2-mm CCI injury resulted in significant spatial learning and memory deficits compared with that in sham adults [11,12]. In previous studies, the Barnes circular maze was shown to be an efficient cognitive task to assess spatial/non-spatial learning following CCI injury in adult mice [11,12]. In addition to cognitive changes, TBI has been frequently linked to affective disorders such as anxiety and depression. However, much remains to be understood about the underlying molecular and signaling mechanisms that mediate affective dysfunctions following injury [24]. The evaluation of anxiety using the OFT and LDT in the present study showed differences in anxiety-like behaviors after CCI injury.
As described above, a growing body of experimental evidence indicates that chronic stress and TBI can lead to cognitive and affective dysfunction, respectively. We showed that juvenile mice exposed to chronic stress would likely suffer from spatial learning and memory dysfunction after TBI during adulthood relative to juvenile mice without chronic stress. In addition, behavioral alterations suggestive of anxiety-like and depression-like behaviors after TBI could be aggravated. It is unclear whether chronic stress and TBI have an overlapping effect on neurobehavioral dysfunction or whether chronic stress increases neuronal injury sensitivity to aggravate neuronal injury following TBI. However, the current study suggests that chronic stress during the juvenile period could contribute to secondary aggravating factors for neurobehavioral deficits following the same traumatic injury impact in adulthood.
To understand whether adolescent chronic stress accelerates the injury process after TBI, which worsens neurobehavioral abnormalities, we evaluated the extent of neuronal damage (loss of the NeuN signal), glial reactions (GFAP for astrocytes and Iba1 for microglia), and apoptotic reactions (Bax/Bcl-2 and procaspase-3). NeuN immunoreactivity has been widely used to identify live mature neurons in brain tissues and measure the neuron/glia ratio in brain regions. Reactive astrogliosis is a key component of cellular response to neuroinflammation. Astrocytic changes were evaluated using an antibody against GFAP, a reactive astrocyte marker. Microglia are the main form of adaptive immune response in the central nervous system, which modulates neuronal function during inflammatory responses and developmental synaptic pruning and plasticity in the healthy brain, and can rapidly respond to even minor changes in the brain [25]. In response to harmful stimuli, microglial cells undergo several changes, such as an increase in the number of proinflammatory cytokines and the expression of several cell surface antigens [25ŌĆō27]. Iba-1 has been widely used to study microglia because its expression is specific and is expressed by both reactive and quiescent microglial cells [27].
Neuroinflammation is a prominent short-term and long-term consequence of neuronal injuries that occur after TBI. It involves the activation of glia, including microglia and astrocytes, to release inflammatory mediators within the brain and subsequent recruitment of peripheral immune cells [28]. Various animal models of TBI have been developed that have proven valuable in elucidating the pathophysiology of the disorder and assessing the safety and efficacy of novel therapies before clinical trials [28]. These studies have reported a robust elevation of cytokines in brain homogenates after TBI [29]. Our data also show that TBI induces microglial and astrocyte reactivity and neural damage in ipsilateral brain homogenates that are altered by chronic stress.
There is consistent evidence that a range of psychosocial stressors during childhood leads to elevated microglial activity and proinflammatory cytokines in the hippocampus and other brain regions [25,30]. Such an elevated neuroinflammatory response may be associated with structural and functional changes in the brain that predispose it to a high risk of mental illness in adulthood. Alterations in the hypothalamic-pituitary stress system, abnormal immunological responses, and lasting changes in cellular, molecular, and epigenetic forms of plasticity have been proposed to explain the neurobiological pathways that link childhood adversities to the later development of adult mental illnesses [25]. Our data also showed that chronic stress induces microgliosis and astrocytosis in brain homogenates.
Our novel finding was that chronic stress enhances microglial and astrocyte reactivity induced by TBI. Diz-Chaves et al. [31] found that prenatal stress induces a basal proinflammatory status in hippocampal formation during adulthood, resulting in the potentiated activation of microglia and astrocytes in response to a subsequent proinflammatory insult. Similarly, another study found that combined exposure to prenatal immune challenge and peripubertal stress induces synergistic pathological effects on adult behavioral functions and neurochemistry, demonstrating that a prenatal insult markedly increases the vulnerability of pubescent offspring to brain immune changes in response to stress [32]. Such studies support the idea that stress leads to microglial priming, whereby an initial stimulation early in life primes microglia, leading to an exaggerated response of microglia to a second inflammatory stimulus [25,33]. Although the findings of the two-hit hypothesis suggest that early life stress primes microglia, leading to a potentiated response to subsequent psychiatric stress, our results suggest that adolescent stress primes microglia, leading to an enhanced response to subsequent nonpsychiatric stress induced by TBI in adulthood.
This study has several limitations. First, we did not evaluate female mice or other mouse strains. Second, we did not analyze time-dependent protein expression early after injury. Third, we impacted the focal right parietal lobe; therefore, we need to further investigate other types of focal and diffuse brain injuries. Finally, we did not administer more than 2.0-mm impact owing to its high mortality rate after surgery.
In summary, our study shows for the first time that chronic stress in the developing brain exacerbates neuroinflammatory responses and neurobehavioral dysfunction in mice exposed to moderate-to-severe TBI that occurs in adulthood. Furthermore, the present study suggests that chronic juvenile stress primes neuroinflammation, leading to enhanced injury response to subsequent TBI.