INTRODUCTION
According to the Global Burden of Disease Study 2019 [1], traumatic injury claimed the lives of 2.4 million people around the world and constituted 7.8% of all deaths. Although head injury and hemorrhage are the two most common causes of death from traumatic injury [2], death due to head injury is reported to decrease with the development of the trauma care system and damage control interventions, whereas death due to hemorrhage does not decrease significantly [3]. In addition, although hemorrhage is the most common cause of early trauma death, up to 50% of patients with traumatic hemorrhage can be potentially saved [3,4].
Traumatic hemorrhage is exacerbated by acute traumatic coagulopathy (ATC). In the past, traumatic coagulopathy was attributed to hemodilution from crystalloid fluid resuscitation, along with progressive hypothermia and acidosis, and was considered an unavoidable consequence of resuscitation. However, Brohi et al. [5] reported that clinically relevant ATC may not be related to fluid administration. They found that patients with evidence of ATC on arrival to the emergency department had significantly higher mortality that was positively correlated with the severity of the injury and not the volume of intravenous fluid administered. Another study by Floccard et al. [6] and Brohi et al. [7] reported that coagulopathy existed at the site of injury in about half the trauma patients managed by mobile intensive care units, and its severity was related to the degrees of injury and hypoperfusion. Thus, if trauma patients experience more severe injuries with significant hemorrhage, hemostasis becomes more challenging. To overcome ATC in severely injured patients, current practices in trauma resuscitation include rapid control of bleeding and maintenance of blood’s hemostatic ability, termed “damage control resuscitation” or “hemostatic resuscitation.” These methods include the use of blood products with very limited use of crystalloid therapy to reduce mortality [8–12].
Timely hemostatic resuscitation using a balanced transfusion ratio of red blood cells (RBCs), plasma, and platelets or whole blood coupled with the use of antifibrinolytic agents such as tranexamic acid has not only improved outcomes, but also decreased inflammatory complications (acute respiratory distress syndrome), number of operations, and the overall amount of used blood products [10]. Thus, hemostatic resuscitation in patients with traumatic hemorrhage appears to give additional protection due in part to its effects on the endothelium, which is injured by hemorrhagic shock [13]. The term “endotheliopathy of trauma (EoT)” was first used by Holcomb and Pati [14] and describes the early damage of the endothelial glycocalyx layer (EGL) after injury. Breakdown of the EGL increases vascular permeability resulting in capillary leakage and exposure of endothelial cells to platelets and white blood cells. These promote thromboinflammation, edema, and organ-barrier dysfunction [15].
ACT and EoT appear to be intricately connected and correlated based on the degree of hypoperfusion. Therefore, this review aims to explore the interconnectedness between these entities.
TRAUMATIC HEMORRHAGE AND OXYGEN DEBT
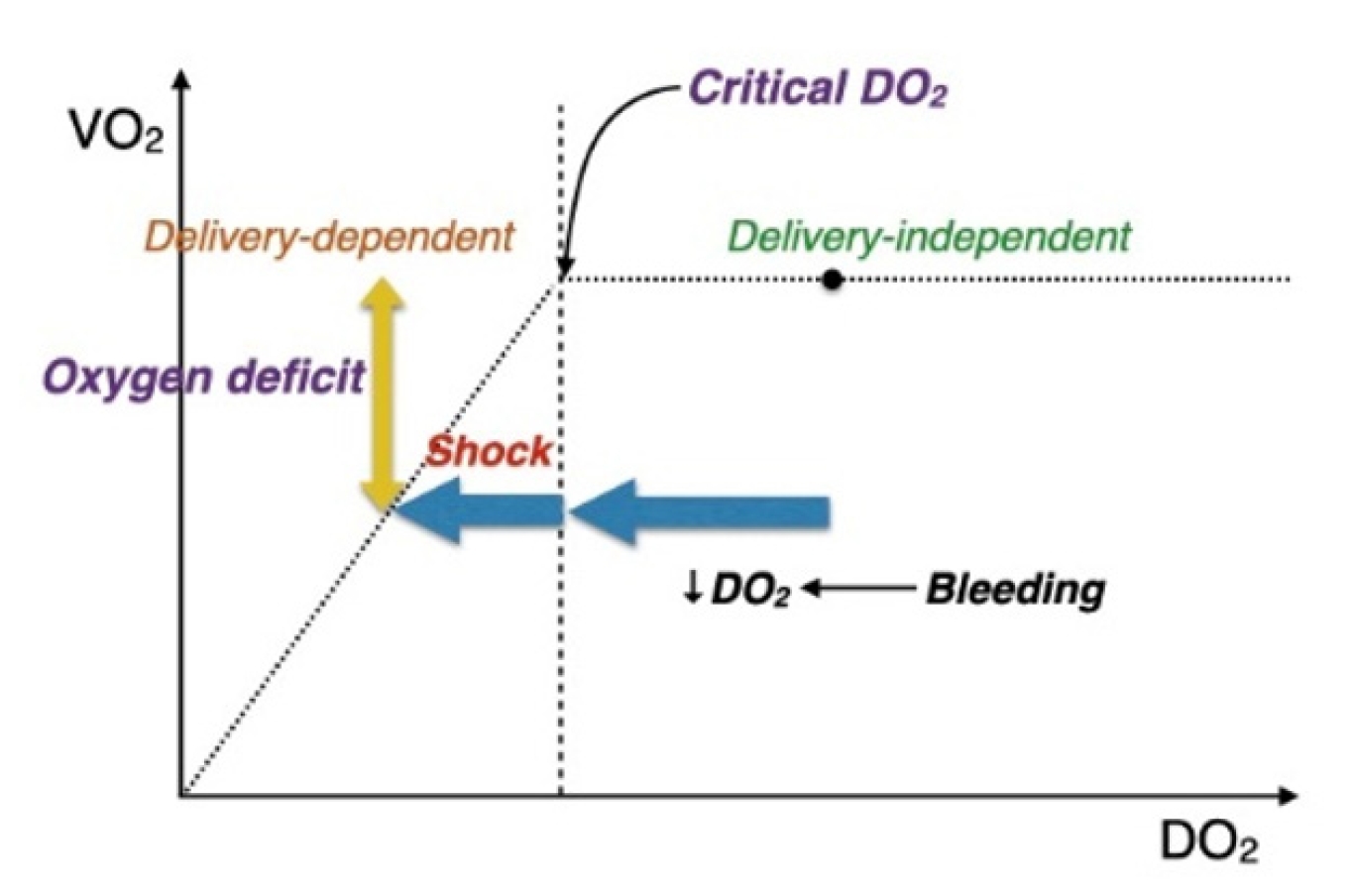
The relationship between oxygen delivery and oxygen consumption is biphasic (Fig. 1). In a normal resting state, metabolic demand and consumption of oxygen (VO2) are independent of oxygen delivery (DO2). This steady state of consumption is achieved by tissues extracting larger amounts of oxygen, resulting in hemoglobin leaving organs with lower levels of oxygen saturation. However, when DO2 decreases below a critical point (critical DO2), the levels of oxygen extraction cannot support aerobic metabolism. At this point, major organ systems enter a state of anaerobic metabolism, and an oxygen deficit occurs. Increase of this level (degree of VO2 below aerobic threshold) over time is the oxygen debt, which can be viewed as whole-body ischemia. Although it is measured precisely in a laboratory setting by indirect calorimetry, oxygen debt can also be reflected clinically (and less precisely) by sequential measures of lactate. Traumatic hemorrhage results in a decrease in DO2 through decreases in cardiac output and blood oxygen content. Additional factors such as vasoconstriction add to a further reduction in DO2 to tissues [16,17]. The resulting oxygen debt quantitatively predicts survival and the development of multiple organ failure after hemorrhage [18–20]. The magnitude of oxygen debt is also linked to levels of mitochondrial dysfunction, oxidative reperfusion injury, and inflammation, leading to necrosis and apoptosis, which contribute to organ dysfunction or failure and even death. The mechanisms of oxygen debt and the resulting cascade of injury also have implications for endothelial damage and coagulation.
Rapid resuscitation from traumatic hemorrhagic shock is critical to not only halt the further accumulation of oxygen debt, but also for its repayment [16]. Failure to repay oxygen debt in a timely manner results in increased reperfusion and inflammatory injury, organ dysfunction, and possible death [21]. However, the ability to ensure repayment is challenging given the biphasic relationship between oxygen delivery and consumption. Stabilizing patients just above critical DO2 will halt anaerobiosis and assist lactate clearance but may not produce the above-baseline levels of VO2 that will result in partial or full repayment. However, early and rapid clearance of lactate appears to be associated with improved chances of oxygen debt repayment.
BLOOD AS AN ORGAN: DEFINITION OF BLOOD FAILURE
A body organ is a structural unit consisting of variable cells that work together in coordinated functions. Blood in the circulatory system is enveloped by the endothelium; the vascular endothelium, as a conduit for blood, coordinates the physiological transport of oxygen, carbon dioxide, nutrients, and waste products and connecting physically distant organs. In addition, the endothelium maintains the fluidity of blood by preventing the formation of blood clots through a thromboresistant surface and anticoagulant mediators that function in the endothelium under normal circumstances. Thus, the blood and the endothelium interact intimately, and the blood-endothelial system can be viewed as an individual organ. In this context, it may be considered the largest integrated functional organ system in the body. As such, the blood-endothelial system is a major target of damage from trauma, hemorrhage, hypoperfusion, and reperfusion injury. Traumatic hemorrhage is capable of inducing endothelial dysfunction and damage, which is believed to begin a cascade of ATC [22,23].
It has been suggested that oxygen debt is the main driver behind ATC [5], and reperfusion injury caused by resuscitation may damage fibrinogen and fibrin [24], weakening clots weaker, exacerbating hemorrhage, and worsening oxygen debt. Thus, oxygen debt, endotheliopathy, and ATC are interconnected and are considered collectively as “blood failure” (Fig. 2). Blood failure can be defined as an emergent state of blood that arises during the accumulation of a critical level of oxygen debt.
When oxygen debt occurs, cellular energetic processes change from aerobic to anaerobic metabolism. Normally, oxygen acts as a terminal electron acceptor in the mitochondrial electron transport chain (ETC). Specifically, glucose and fatty acids are metabolized in the cytoplasm and mitochondria, respectively, generating high-energy electron carriers, such as NADH and FADH2. These electron carriers donate their electrons to the ETC, where they pass through a series of protein complexes, including complexes I, II, and III. Finally, at complex IV, molecular oxygen (O2) serves as the final electron acceptor. At this stage, oxygen accepts the electrons donated by the electron carriers and, together with protons (H+) from the mitochondrial matrix, forms water (H2O). This electron transfer reaction allows the flow of electrons to continue within the ETC, producing the proton gradient that drives the synthesis of adenosine triphosphate (ATP) through ATP synthase. The use of oxygen as the terminal electron acceptor in complex IV is crucial for efficient production of ATP, providing the cell with a continuous and robust energy supply to support essential functions. However, under conditions of critically low oxygen delivery, the ETC may leak electrons, leading to the production of reactive oxygen species (ROS). These ROS, including superoxide anion and hydrogen peroxide, can damage cellular components, such as lipids, proteins, and DNA. Electron leaks occur when electrons prematurely escape the ETC and react with oxygen, bypassing complex IV. This uncoupling of electron flow from ATP synthesis disrupts energy production, exacerbates cellular stress, and contributes to mitochondrial dysfunction. In addition, lactate accumulates as a byproduct when the rate of glycolysis exceeds the capacity of oxidative metabolism to utilize pyruvate. This process helps sustain energy production in cells but leads to lactate accumulation, which can decrease pH and metabolic acidosis. Moreover, neurovascular compensation through sympathetic activation and catecholamine release activates Na+/K+-ATPase, increasing lactate production [25] and constricting blood vessels to further reduce blood flow to organs and increase tissue hypoxia. Changes in blood redox potential, accumulation of lactic acid, and an increase of catecholamine resulting from the accumulation of oxygen debt affect endothelial cells and blood components and cause EoT and ATC, which represent blood failure [26–28].
ENDOTHELIOPATHY OF TRAUMA
The endothelium is one of the largest parts of the human body, occupying a blood-endothelial interface measuring approximately 300 to 1,000 m2 [29]. The endothelium is formed by a single cell layer and lines all blood and lymphatic vessels. The physiological function of the endothelium includes control of vascular tone, inflammation, angiogenesis, and regulation of coagulation and fibrinolysis [14,30,31]. The endothelial glycocalyx lines the inner surface of the vascular endothelium and consists of membrane-bound proteoglycans, glycoproteins, and soluble plasma or endothelium-derived molecules [32]. When traumatic hemorrhage-induced oxygen debt occurs, it results in hypoxia and catecholamine release. In addition, release of inflammatory mediators and enzymes such as matrix metalloproteinases and heparanases from injured tissue and leukocytes induce shedding of the glycocalyx [22,33,34]. This shedding from the endothelial surface results in loss of barrier function, leading to capillary leaks; exposure of heparan sulfate from endothelial cells, which contributes to coagulopathy through autoheparinization; and release of damage-associated molecular patterns, contributing to systemic inflammation [35–38]. Glycocalyx shedding also exposes the endothelial cell surface and initiates nonspecific adhesion of platelets and leukocytes and generation of thrombin [39,40]. In addition to glycocalyx shedding, ROS and circulating proinflammatory cytokines such as TNF-α and interleukin 6 (IL-6) induce endothelial cells to propagate innate immune responses [41,42]. This shedding of the endothelial glycocalyx and endothelial cell activation result in capillary leaks, nonspecific intravascular coagulation, and inflammation throughout the systemic endothelium of the body. In other words, this is not a localized effect at the site of a traumatic injury.
ACUTE TRAUMATIC COAGULOPATHY
Coagulopathy after traumatic hemorrhage has long been acknowledged and considered a co-phenomenon and inevitable consequence of large-volume crystalloid resuscitation, hypothermia, and metabolic acidosis. However, endogenous coagulopathy caused by the severity of the traumatic injury itself and the degree of traumatic shock has been recognized and named ATC [5,47]. ATC is a complex and multifactorial condition that begins in the early stages of traumatic hemorrhage and can rapidly progress as level of oxygen debt increases. Several mechanisms of ATC have been proposed, with additional proposals under investigation. Despite this, it is agreed that ATC involves disturbances in platelet function, fibrinogen depletion, and dysregulated fibrinolysis, leading to impaired blood clotting, excessive bleeding, and potential accumulation of greater oxygen debt.
Platelet dysfunction is a critical aspect of ATC. Trauma-induced shock and tissue injury can activate platelets, causing them to aggregate and release procoagulant substances. However, platelets may also become dysfunctional, leading to decreased clot formation and impaired hemostasis. In a swine traumatic hemorrhage model, it was demonstrated that clot strength is reduced as oxygen debt increases despite unchanged platelet count [48]. In addition, inhibition of platelet function eliminated increases in the firmness of clots in a rat polytrauma model [49]. These findings suggest the importance of platelet function in clot strength, and platelet dysfunction results in weak clots. It has also been reported that platelets from trauma patients show impaired aggregation to ex vivo agonist stimulation independent of platelet count, and this is more pronounced in nonsurvivors [50,51]. Impaired aggregation of platelets was shown to develop very quickly in a porcine model of traumatic hemorrhage and can occur within 15 minutes of injury [52]. The precise mechanism is not known but involves the endothelial release of tissue factor, platelet-activating factor, and von Willebrand factor [53,54], which activates platelets beyond their primary role of hemostasis. This phenomenon is referred to as “platelet exhaustion” [51,55,56]. Circulating soluble factors, which remain undefined, are thought to mediate platelet exhaustion [57], and a platelet transfusion may not necessarily reverse platelet dysfunction [58,59]. In addition to impaired platelet aggregation in patients with traumatic hemorrhage, impairments in the adhesive function of platelets to collagen and contractile force have been reported [60,61]. Platelets with an impaired aggregation response also contribute to tissue plasminogen activator (tPA)-mediated fibrinolysis due to impaired platelet plasminogen activator inhibitor-1 (PAI-1) [62].
Poor survival has been reported in trauma patients with diminished level of fibrinogen [63]. In a swine model of trauma and hemorrhage, fibrinogen was significantly reduced with increasing oxygen debt [48]. Fibrinogen is converted to fibrin by thrombin and forms a hemostatic plug, together with platelets. It is the terminal substrate for the coagulation cascade and must be maintained at a minimum level for effective hemostasis. The mechanisms for diminished level of fibrinogen in traumatic hemorrhage include impairment of fibrinogen synthesis due to hypothermia, accelerated degradation by acidosis and reperfusion injury, and consumption in clot formation with additional blood loss and hemodilution, further decreasing fibrinogen level [64–66].
Dysregulated fibrinolysis may also be involved in ATC, but the exact mechanism in patients with traumatic hemorrhage remains unclear. Exposure of tissue factors by injury and endothelial disruption activates the extrinsic coagulation pathway and generates thrombin, leading to fibrin and clot formation. Initially, the clotting process is localized at the site of injury, but escape of thrombin from the injury site activates the systemic coagulation process [67]. Normally, thrombin escaped from the injury site is inhibited by circulating antithrombin and thrombomodulin expressed on endothelial cells, and the thrombin-thrombomodulin complex activates protein C to maintain tissue perfusion by inhibiting thrombosis. However, persistent tissue hypoperfusion induces endothelial cell damage, which increases the levels of soluble thrombomodulin and the thrombin-thrombomodulin complex to excessively activate protein C and interfere with blood coagulation [67]. It has been reported that soluble thrombomodulin and activated protein C were increased in trauma patients and are associated with poor clinical outcomes [68]. Although activated protein C is involved in control of fibrinolysis by cleavage of factors Va and VIIIa, as well as binding of PAI-1, and is suggested as a major driver of ATC, the level of activated protein C in trauma patients cannot cleave factor Va efficiently [69]. Rather, hyperfibrinolysis observed in ATC might be associated with the overwhelming systemic release of tissue plasminogen activator from Weibel-Palade bodies stored in endothelial cells and loss of alpha2 antiplasmin and PAI-1 [70–75]. Another concept of dysregulated fibrinolysis is termed “fibrinolysis shutdown” and has been reported as the most common phenotype of dysregulated fibrinolysis in severe trauma patients [76]. Patients who exhibited fibrinolysis shutdown experienced prior hyperfibrinolysis, including elevation of D-dimers and depletion of fibrinolytic inhibitors [77]. The mechanism of fibrinolysis shutdown is not fully identified. A surge in PAI-1, shedding of S100A10 plasminogen receptor protein from the surface of endothelial cells that normally drive the hyperfibrinolysis process, and increased level of thrombin-activatable fibrinolysis inhibitor in plasma have been suggested [78–80]. The extent of these processes and level of fibrinolysis are greatest in patients with rapid and massive hemorrhage and reflected in higher oxygen debt [62,76].
CONSIDERATIONS IN THE MANAGEMENT OF BLOOD FAILURE
As indicated above, severe traumatic hemorrhage can result in blood failure, which is collectively composed of oxygen debt, EoT, and ATC. Increasing levels of EoT and ATC will complicate hemostasis, leading to accumulation of greater oxygen debt. To prevent or reverse blood failure in patients with traumatic hemorrhage, immediate control of hemorrhage and repayment of oxygen debt must be combined with simultaneous treatment of both endothelial injury and coagulopathy. Damage control resuscitation (DCR) or hemostatic resuscitation is a strategic approach in patients with severe injuries and hemorrhagic shock. DCR includes immediate control of ongoing hemorrhage, early balanced transfusion of blood products with other hemostatic agents, and permissive hypotension. The combination and simultaneous execution of these strategies reduce the degree of blood failure.
Transfusion of packed RBCs (pRBCs) increases the oxygen-carrying capacity, cardiac output, and thus DO2. In addition, RBCs influence hemostasis as evidenced by increased accumulation of platelets at a high hematocrit level and the association of increased RBC count with predisposition to thrombosis [81,82]. The proposed mechanisms include increased platelet adhesion and aggregation through the release of adenosine diphosphate and thromboxane A2, the release of membrane-derived procoagulant microvesicles, and aggregation of RBCs with platelets via adhesive molecules [83]. Thus, early use of pRBCs can help to decrease the oxygen debt and provide additional hemostatic effects. There has been concern that pRBCs stored a long period might have altered oxygen affinity and delivery, rheological changes, and adhesiveness to the endothelium, which might affect outcomes [84–86]. However, a recent multicenter, randomized blinded trial did not show any benefit of fresh RBCs in critically ill patients, although most of the patients were not victims of trauma [87,88].
Most guidelines recommend the transfusion of a high ratio of plasma to RBCs, with the majority recommending a 1:1 ratio. Transfusion of plasma increases cardiac output through intravascular volume expansion and supplements coagulation factors to increase hemostatic ability. Additionally, plasma may provide endothelial protection, assisting in recovery of the endothelial glycocalyx, abrogating endothelial hyperpermeability and inflammation, and restoring syndecan-1 expression in the lungs [89–92]. Plasma also promotes homeostasis in thrombin generation [93]. Considering that EoT and ATC develop rapidly after injury, prehospital plasma transfusion may be helpful. Based on this, several militaries have invested in the development and deployment of lyophilized plasma that can be stored, carried, and reconstituted in field environments. However, controversial reports exist on the benefits of prehospital administration of plasma alone on survival and may reflect the need for additional components (pRBCs and platelets) during longer transport times [94,95]. While minor, transfusion of plasma has risks including infectious disease transmission and triggering of transfusion-related acute lung injury [96,97].
Early platelet transfusion in conjunction with pRBCs (1:1 ratio) is associated with improved outcomes in traumatic hemorrhage [98]. Platelets contribute to hemostasis by increasing thrombin formation, clot firmness, and resistance to clot lysis [99]. However, platelets have a limited shelf-life, compromising immediate availability. Platelets are traditionally stored at room temperature to maintain function, but storage is usually limited to 5 days due to growth of bacterial contaminants. However, evidence suggests that storage of platelets at 4 °C may extend the effective lifespan for up to 14 days and may decrease the risk of infectious complications [100–102]. Although cold-stored platelets remain in circulation for a shorter period, they showed better hemostatic function and greater capacity to inhibit endothelial permeability than platelets stored at room temperature [101,102].
Fresh and cold-stored whole blood is a growing alternative to the 1:1:1 ratio of pRBCs, plasma, and platelets. The use of whole blood can not only restore oxygen-carrying capacity, but also mitigate endotheliopathy and coagulopathy simultaneously and simplify the logistics of massive transfusion protocols. The use of whole blood has been limited to military medicine, but its use has been increasing recently in civilian trauma including several prehospital ground and air ambulance systems [103–106]. Although no large randomized control trial currently exists, the use of whole blood has been reported as feasible and associated with improved survival in several studies [107–111].
Transfusion of cryoprecipitate, a rich source of fibrinogen, factor VIII, and von Willebrand factor, and fibrinogen concentrate can be used for supplementing fibrinogen and enhancing hemostasis. A minimal level of fibrinogen is required to maintain effective hemostasis. It has been reported that the use of fibrinogen concentrates or cryoprecipitate in trauma patients resulted in better survival [112–117]. However, there are controversies surrounding the effect of fibrinogen supplements on traumatic hemorrhage. A meta-analysis that included four randomized controlled trials [118] found that fibrinogen concentrate had no significant benefit on mortality, although the quality of evidence was graded low to moderate.
Current practice standards for DCR recommend early administration of tranexamic acid (TXA) for facilitating hemostasis. The CRASH-2 (Clinical Randomisation of an Antifibrinolytic in Significant Haemorrhage 2) trial [119] demonstrated that administration of TXA within 3 hours of injury significantly reduced mortality, especially in hypotensive patients. Although a recent study [120] comparing TXA and placebo within 2 hours of injury in prehospital patients with hypotension (systolic blood pressure, <90 mmHg) and tachycardia (heart rate, >110 beats/min) did not result in significantly lower mortality, a subgroup analysis of patients with severe shock (systolic blood pressure, <70 mmHg) or TXA administered within 1 hour of injury showed a survival benefit. Thus, TXA may be beneficial only in patients with severe shock and should be used shortly after the trauma event. The potential risk of TXA treatment is the development of venous thromboembolism (VTE), as demonstrated in retrospective studies of trauma patients [121,122]. Administration of TXA may increase the incidence of fibrinolysis shutdown, and patients in the CRASH-2 trial who received TXA at 3 hours after injury had an increased risk of death [66,123]. In addition, a small, single-center, randomized trial [124] compared placebo with 2 or 4 g TXA administration within 2 hours of trauma in patients requiring a transfusion of at least 1 unit of RBC and showed a dose-dependent increase of thromboembolic events. Furthermore, a large study administering TXA continuously for 24 hours in patients with acute gastrointestinal bleeding [125] showed a higher incidence of VTE. Thus, the use of TXA should be guided by coagulation status in patients. Conventional coagulation tests, such as prothrombin time, activated partial thromboplastin time, fibrinogen level, and platelet count, are unable to identify the status of hyperfibrinolysis or hypofibrinolysis. However, viscoelastic hemostatic assays can provide more comprehensive information about the coagulation status of patients rapidly and in real-time [22,126].
Permissive hypotension before and during definite bleeding control is advocated to limit ongoing hemorrhage by reducing hydrostatic pressure while maintaining a level of critical vital organ perfusion [127–132]. During permissive hypotension, providers control resuscitation so that systolic blood pressure does not exceed a targeted pressure (e.g., 100–110 mmHg). This strategy may reduce the degree of noncompressible torso hemorrhage until definitive surgical hemostasis can be achieved. However, prolonged definite bleeding control can produce permissive hypotension to result in additional accumulation of oxygen debt and must be balanced. Such strategies are even more complicated in patients in shock who also have traumatic brain injuries, in whom the recommended minimal systolic blood pressure is 110 to 120 mmHg [133,134]. The balance between permissive hypotension and avoiding the accumulation of additional oxygen debt is not trivial. The use of blood product resuscitation will help maximize the potential benefit of permissive hypotension.
CONCLUSION
Blood and the vascular endothelium can be considered a single organ system that can fail because of oxygen debt incurred during traumatic hemorrhage and its repair. The severity of this failure is exhibited through the development of EoT and ATC. This interplay among oxygen debt, EoT, and ATC drives the concept of "blood failure," where the disruption of normal physiological homeostasis results in a cascade of negative effects.
Efforts in managing blood failure involve a multifaceted approach. DCR strategies, such as permissive hypotension, early control of hemorrhage, and balanced transfusion of blood products, aim to limit the accumulation of oxygen debt and maximize its repayment, alleviate endothelial damage, and restore coagulation. Transfusion of pRBCs, plasma, and platelets and the use of TXA are essential components of DCR, contributing to hemostasis and mitigating the effects of ATC. Additionally, the use of whole blood, whether fresh or cold-stored, is emerging as a promising approach to address the holistic needs of trauma patients. The intricate relationships between these factors highlight the urgent need for comprehensive, integrated strategies that consider the interconnected nature of traumatic hemorrhage, blood failure, and its associated complications. This review sheds light on synergistic relationships and emphasizes the importance of multidisciplinary approaches to effectively manage traumatic hemorrhage and to mitigate its detrimental consequences.