INTRODUCTION
According to the World Health Organization and epidemiological data, traumatic brain injury (TBI) is a significant global public health issue and a major cause of morbidity and mortality [1]. In the United States, TBI affects more than 1.5 million people annually, out of which approximately 300,000 are hospitalized and more than 50,000 deaths, accounting for approximately one-third of injury-related deaths [2,3].
In addition to primary brain injuries, secondary insults such as hypoxia, hypotension, hypercapnia, and seizures can occur and further aggravate the injury [4]. Hypoxia is a frequent secondary insult correlated with poor outcomes [5]. Cerebral hypoxia has been reported in up to 44% of TBI patients with underlying brainstem injury, hypotension, impaired brain perfusion, and extracranial trauma, including airway obstruction, chest injury, and peripheral hemorrhage [6].
TBI may result in various temporary or permanent physical, cognitive, behavioral, and emotional impairments [7]. Among these, initial and persistent cognitive deficits are the most common, including loss of short-term memory and learning difficulties. The pathophysiology of TBI-induced neurobehavioral changes is underpinned by biochemical processes that lead to secondary brain damage, such as neurotoxic end product release, neuroinflammation, formation of reactive oxygen species (ROS), and neuronal cell death [4,6,8].
Although large epidemiological studies have reported that postTBI hypoxia exacerbates neurobehavioral deficits, experimental studies using murine models examining such effects are limited. Recent studies using the Marmarou rat model of diffuse TBI revealed that secondary systemic hypoxia aggravates behavioral deficits, enhances neuroinflammation, and causes prolonged metabolic dysfunction [9,10]. Murine fluid percussion and controlled cortical impact (CCI) models of focal TBI demonstrated that secondary systemic hypoxic insults worsen functional outcomes and increase neuronal cell death [11]. These post-TBI systemic hypoxia models, in which oxygen delivery is strictly controlled throughout the experimental procedure, well reflect the hypoxic conditions caused by an obstruction of the airways or other respiratory difficulties following an accident. However, there is a paucity in experimental studies combining the CCI mouse model with additional posttraumatic hypoxic ischemia (HI). Among TBI animal models, the CCI model produces the most precise injury by controlling deformation parameters, such as time, velocity, and depth of impact. Consequently, its most crucial advantages over other TBI models are the absence of risk for rebound injury as well as higher accuracy, reproducibility, and simplicity [12].
Although there is limited clinical evidence of the neurological impact secondary HI has on TBI [5,13], it is considered a critical factor that augments trauma-induced brain damage. However, the underlying mechanisms responsible for this effect remain inconclusive.
Here, we assessed the differences in neurobehavioral deficits, neuroinflammation, and oxidative stress between TBI and TBI combined with secondary HI (TBI+HI) in adult male mice exposed to moderate to severe CCI.
METHODS
Animal population
All experimental protocols were approved by the Animal Care and Use Committee at Chungnam and Chungbuk National Universities (No. 00-854-15) and adhered to the Declaration of Helsinki. A total of 88 C57Bl/6 male mice aged 8–12 weeks were used in this study. Animals were housed in a standard 12-hour light/dark cycle with ad libitum access to food and water. The numbers of mice used were as follows: preliminary experiments to determine injury severity according to impact depth: 20; neurobehavioral testing: 41, of which 32 were euthanized following tiletamine/zolazepam anesthesia on day 21 after injury; biochemical test: 27, of which 23 were euthanized on day 3 after injury.
Surgical procedures
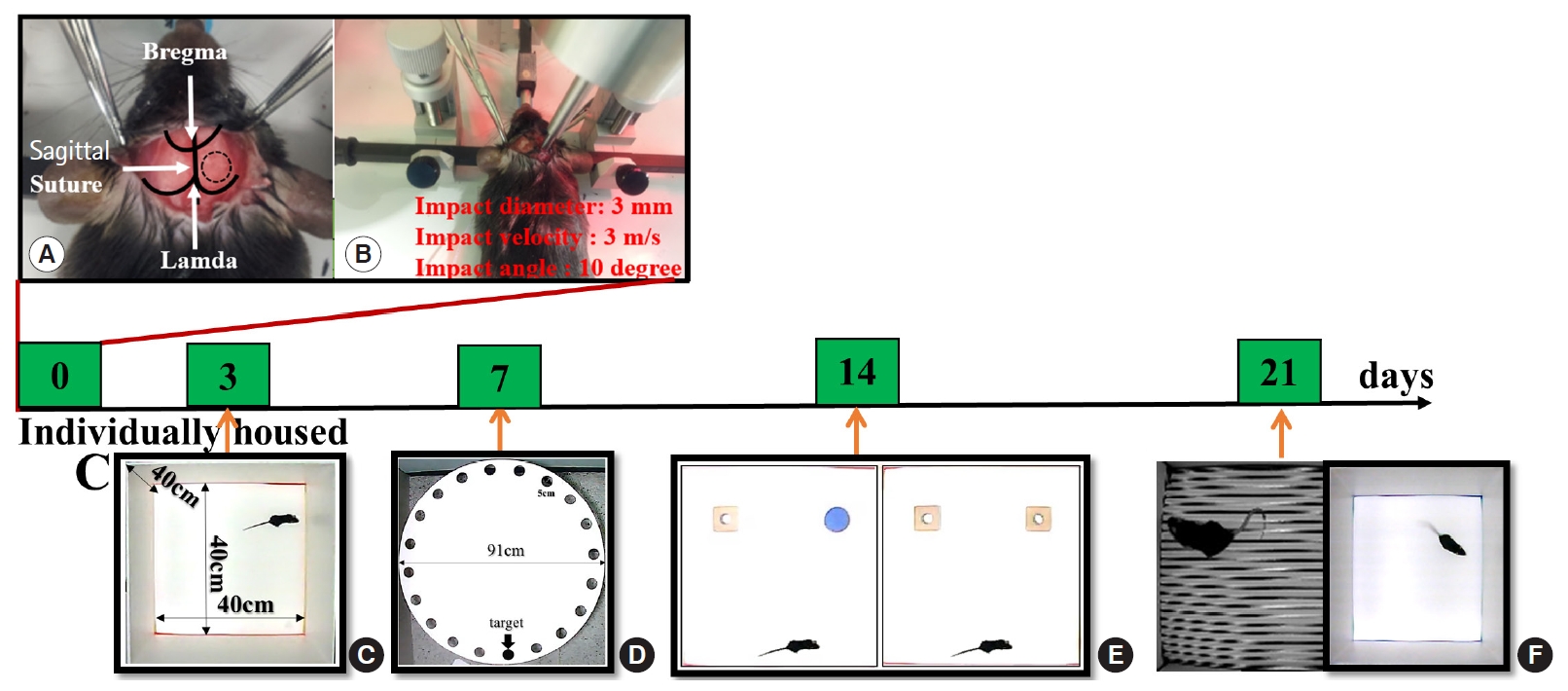
Mice were anesthetized with an intramuscular injection of 15 mg/kg of tiletamine/zolazepam (Zoletil; Virbac, Carros, France). After the scalp was shaved and cleaned with betadine, a midline incision was made to expose the right parietal bone. A 4-mm-diameter circle was drawn at the center of lambda and bregma 0.5 mm from the midline. Right parietal craniotomy was performed using a surgical microscope and micromotor drill (Stoelting, Wood Dale, IL, USA) along the marked circle. The CCI device was calibrated with respect to the exposed dura mater within the craniotomy. Impact parameters for injury comprised a depth of 0.0, 1.0, 1.5, and 2.0 mm, mean velocity of 3.0 m/s, and duration of 500 ms. Following the impact, the scalp incision was sutured with 5-0 nylon. After supine positioning we induced unilateral cerebral HI damage as described previously [14,15]. The right carotid artery was carefully isolated without damaging the vagus nerve and double ligated with 4.0 surgical silk prior to suturing. Animals were allowed to recover with access to food and water for 2 hours before exposure to hypoxia in hypoxic chambers for 30 minutes. Oxygen levels were monitored and kept at 8%, balanced with nitrogen. Chamber temperature was maintained at 35°C. Animals were returned to their cages post procedure. Sham-operated groups underwent craniotomy without CCI injury. The surgical procedure is shown in Fig. 1.
Health assessment score
The mice were weighed daily. A health assessment score was recorded for each mouse daily postsurgery, ranging from 0–2, 0–3, or 0–5 depending on the behavior assessed (0 indicating no deficit, 5 indicating the most severe impairment). Assessed behaviors included consciousness (0–3), interaction (0–2), ability to grasp a wire top (0–2), motor function (0–5), and activity (0–2) [16,17]. Sums of scores in each category yielded the overall health assessment score.
Hematoxylin-eosin staining and estimation of cortical lesion volume
Animals were anesthetized as described above and perfused with cold saline on day 21 after injury. Brains were removed, post-fixed with 10% formalin, and paraffin embedded. Coronal sections (6-µm thick) were collected at 100-µm interval and stained with hematoxylin and eosin (H&E). Micrographs of H&E-stained sections were obtained to record and calculate the extent of injury at 1.25× magnification using a CX43 Olympus microscope (Olympus Corporation, Toyko, Japan).
Cortical lesion volumes (mm3) were determined by calculating the area of the lesion (mm2) (at 1.25× magnification using the CX43 microscope) by outlining the missing cortical tissue for each section cut at 500-µm intervals and multiplying the sum of lesion areas obtained from each section with the distance between sections. Lesion volumes were analyzed using Image J 1.42 (National Institutes of Health, Bethesda, MD, USA).
Behavioral testing
Behavioral testing was conducted between 8 a.m. and 7 p.m. by an observer blinded to the experimental procedures. Memory was assessed using the Barnes maze test (BMT), contextual and cued fear conditioning (CCFC), and novel object recognition test (NORT) on days 7, 14, and 21 after CCI, respectively (Fig. 1). All testing was recorded using a video tracking system with SMART ver. 3.0 (Harvard Apparatus, Holliston, MA, USA), which automatically identified post-injury behavioral changes.
Locomotor activity
Locomotor activity was measured in a white open-top acrylic box (40×40×40 cm) with an illumination intensity of 20 lux at floor level for 30 minutes. Distance moved (mm), time spent within the center (central 25% of box) and the area surrounding it (sec), and mean walking speed (cm/sec) were evaluated.
BMT
The BMT was conducted as previously described with minor modifications [18]. The maze consisted of a white, acrylic, circular platform (91 cm diameter) with 20 equally sized holes and a black acrylic escape box (20×5×6 cm) along the perimeter. The maze was surrounded by four spatial cues at the height of the maze.
Acquisition trials. Acquisition comprised four trials per day over 3 days with 10–15 minutes inter-trial intervals. Prior to the first trial, the mice were placed in the middle of the maze in a black starting cylinder (10 cm diameter). A loud white noise (80– 90 dB) was sounded, inducing escape behavior. After 10 seconds, the chamber was lifted, and the mice were pretrained to enter the escape tunnel by guiding them to it and allowing them to remain there for 2 minutes. Following this training, the first trial was initiated.
At the start of each trial, the mice were placed in the black starting cylinder. Ten seconds after the onset of the noise and light, the cylinder was lifted and the animals were free to explore the maze. The trial ended when the mice entered the escape tunnel or after 3 minutes. Immediately after the mice entered the tunnel, the noise was terminated. Animals were allowed to stay within the tunnel for 1 minute. After each trial, the maze was cleaned with 70% alcohol and rotated to eliminate intra-maze cues.
Probe trial. During the 90-second probe trial conducted on day 6, the escape tunnel was closed. Mice were allowed to explore the maze and visit the escape tunnel. Primary latency and distance traveled to reach the tunnel were recorded.
CCFC
To assess fear learning and memory, the CCFC test was conducted as previously described with minor modifications [19]. The apparatus consisted of a square chamber (25×25×24 cm) with an electrifiable grid floor, sound source, and calibrated shock generator (Panlab/Harvard Apparatus, Barcelona, Spain).
Conditioning. Animals were placed into the conditioning chamber. After 2 minutes of exploration, an 80-dB clicker (6 cps) sounded for 30 seconds to serve as a conditioned stimulus (CS) and was concomitantly terminated with a 2-second, 0.7-mA scrambled foot shock (unconditioned stimulus, US). This sequence was repeated twice, followed by a 30-second delay before mice were returned to their home cages.
Context test. After completing the conditioning session, the mice were returned to the same conditioning chamber and scored for freezing behavior to measure contextually conditioned fear. A 72-hour delay interval between the conditioning and context test was set. Mice were placed in the conditioning chamber and allowed to freely explore the chamber for 5 minutes without CS and US presentations prior to being returned to their home cages.
Cued test. The cued test was conducted on the same day as the context test. Mice were placed into a different testing chamber for 3 minutes, providing a new context unrelated to the conditioning chamber. At the end of the initial 3 minutes, a conditioned auditory cue was presented for another 3 minutes in the novel context environment. Mice were allowed to explore the chamber for 6 minutes; during the first 3 minutes, neither CS nor US was presented, while a CS (55 dB white noise) was presented during the final 3 minutes.
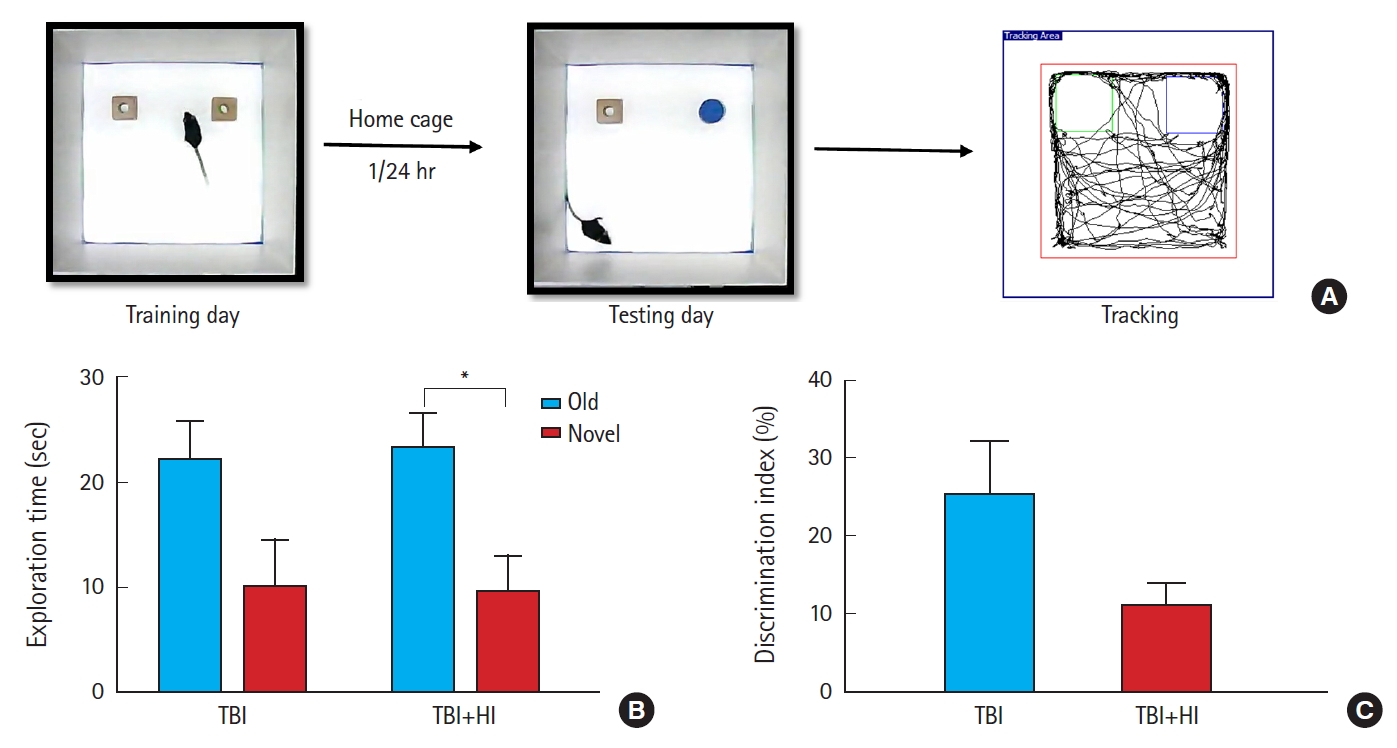
NORT
Behavior was sequentially assessed in the NORT using a 2-day protocol in the same open top acrylic box described above [20]. After habituation, animals were allowed to explore two identical objects for 10 minutes (acquisition trial). The time spent exploring each object was recorded. Retention trials were performed at 1-and 24-hour intervals following the acquisition trial. During retention trials, the mice were returned to the same box, in which one of the familiar objects used during acquisition was replaced by a novel object. Mice were allowed to explore freely for 10 minutes. Time spent exploring each object was recorded.
Measurement of oxidative stress markers
We measured lipid peroxidation, glutathione (GSH) concentration, and superoxide dismutase (SOD) activity as oxidative stress markers on day 3 after injury. Snap-frozen cortex was homogenized with ultrasonic waves in a bead homogenizer. Cortical lipid peroxide content was determined using the modified thiobarbituric acid (TBA)-reactive substances assay method as described previously [21]. Briefly, an aliquot of the homogenate was added to an equal volume of 0.67% (w/v) TBA. Subsequently, 800 μL of the supernatant were mixed with 100 μL of 0.1 M TBA and heated at 100°C for 10 minutes. After cooling, the absorbance was read at 532 nm using a SpectraMax 250 microplate reader (Molecular Devices, Sunnyvale, CA, USA). A malondialdehyde (MDA) standard was prepared from 1,1,3,3-tetraethoxypropane. GSH levels and SOD activity were determined using the GSH colorimetric assay kit (BioVision, Milpitas, CA, USA) and SOD assay kit (Cell Biolabs, CA, USA), respectively, according to the manufacturer’s instructions.
Measurement of proinflammatory cytokines
On 3 day after injury, interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α levels were measured in the supernatant of homogenized brain tissue using a mouse multiplex cytokine assay kit (Multi Beads Assay BioPlex; R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. Cytokine levels were calculated according to multiplex cytokine software.
Statistical analysis
All data are presented as arithmetic mean±standard error of the mean. The statistical significance among groups was assessed with nonparametric Kruskal-Wallis one-way analysis of variance, followed by multiple comparisons post hoc analyses with Bonferroni methods. P<0.05 was considered statistically significant. Data were analyzed using PASW Statistics ver. 18 (SPSS Inc., Chicago, IL, USA).
RESULTS
Physiological variables
Body weight, body temperature during surgery, age, rates of survival, and health assessment scores are shown in Table 1. Weight gain (%) was significantly lower in the TBI+HI group as compared to the TBI-only group on postoperative days (POD) 2−3. In contrast, health assessment scores were significantly enhanced in TBI+HI mice as compared to TBI-only counterparts on POD 1−3. The rate of survival of TBI+HI mice was significantly lower than that of TBI-only mice (Table 1).
Cortical impact injury volumes
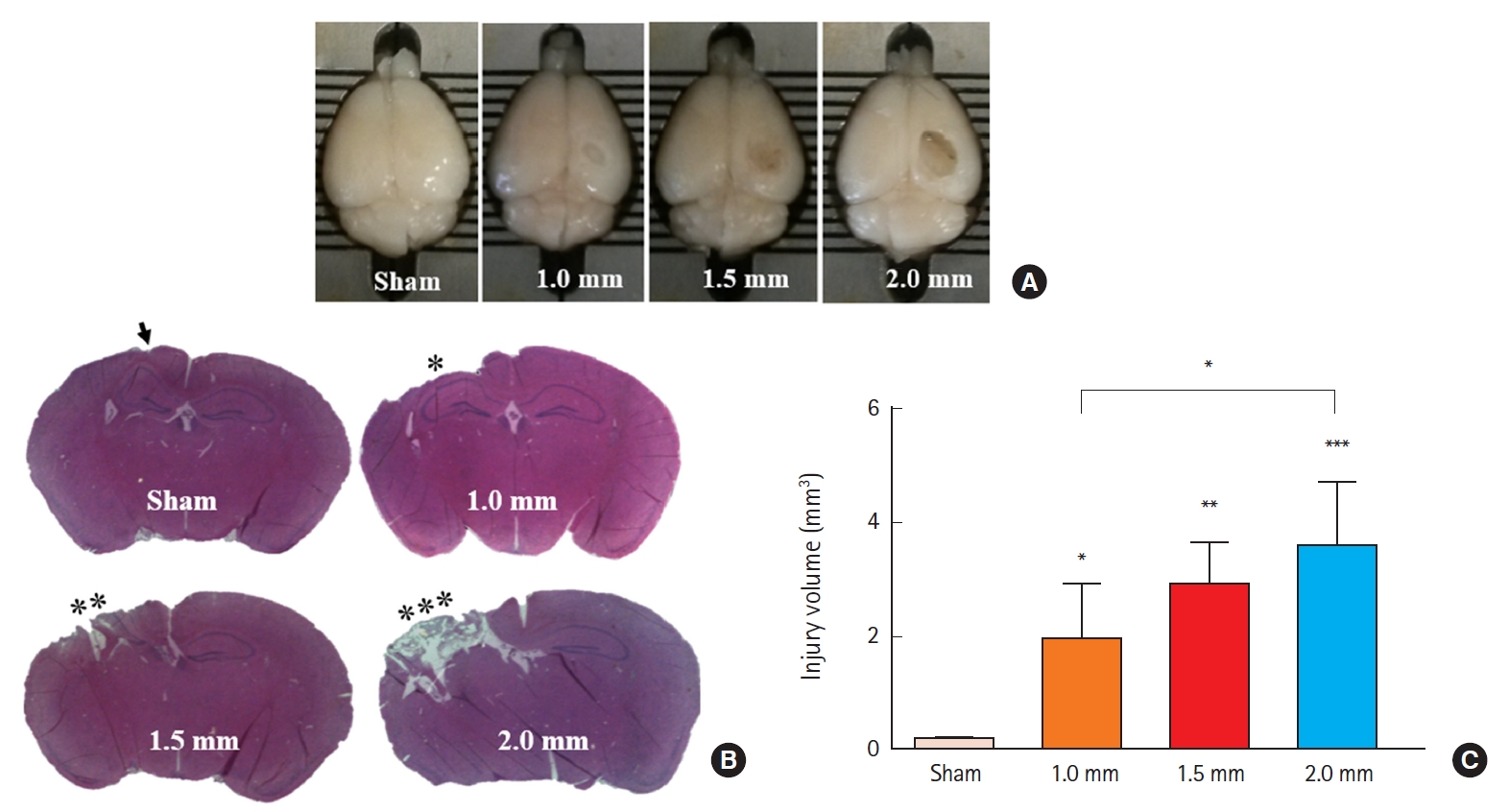
To determine injury severity according to impact depth, we first impacted mouse cortices with four different depths (0.0, 1.0, 1.5, and 2.0 mm) (Fig. 2A). Analysis of tissue samples collected 7 days post-injury demonstrated progressively larger brain lesions proportional to the graded depth of impact. Sham mice were not or minimally injured due to drilling. H&E staining demonstrated that all mice exposed to 2.0-mm impact depth revealed the apparent hippocampal injury, whereas 1.0-mm impact depth did not affect hippocampi (Fig. 2B). Based on the observed differences in injury volume between tested depths of cortical impact (Fig. 2C), we applied 2.0-mm depth of impact (moderate to severe injury) to analyze the differences between TBI-only, TBI+HI, and control groups in subsequent experiments.
Neurobehavioral outcomes
Locomotor activity
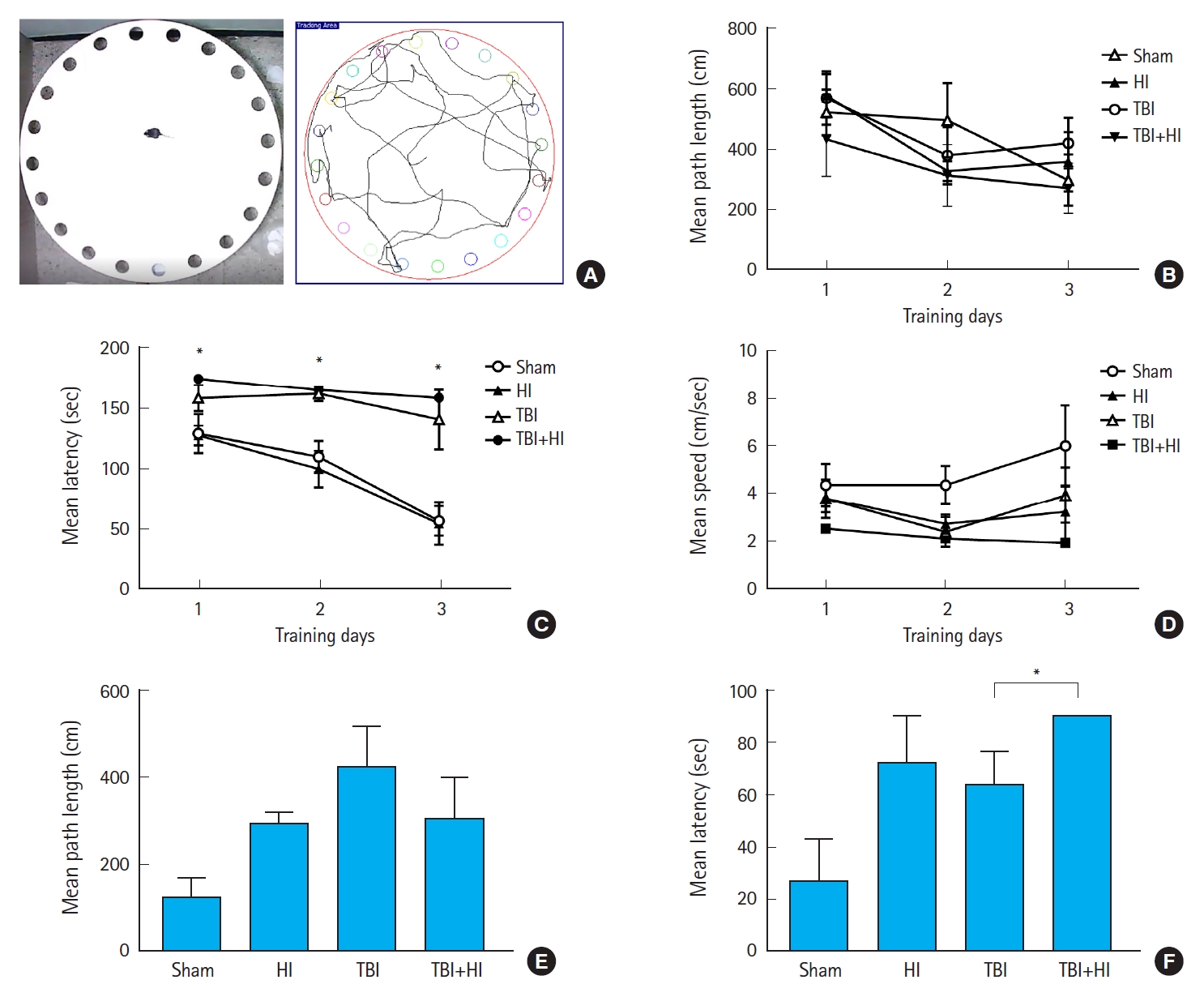
As shown in Fig. 3, overall locomotion was not influenced by TBI or HI, indicating that neither significantly impacted motor behavior.
Spatial learning and memory deficits in the BMT
We next assessed spatial learning and memory using the BMT (Fig. 4A). In the acquisition phase, while there was no change in mean path length among groups (Fig. 4B), the latency to enter the escape tunnel decreased significantly over the three training days in sham mice, indicating mean performance improvement (Fig. 4C). A significant difference was observed in the latency to enter the escape hole between TBI and TBI+HI, and between sham and TBI+HI mice (Fig. 4C). No significant difference was observed in average speed among groups (Fig. 4D). In the retention phase, no significant differences were observed in primary path length to the target hole among groups (Fig. 4E). In contrast, an unpaired t-test revealed significant differences in primary latency to enter the target hole and adjacent holes between TBI and TBI+HI mice, and between sham and TBI mice (Fig. 4F). These findings indicate that TBI caused retention memory deficits that were exacerbated by HI (Fig. 4F).
CCFC
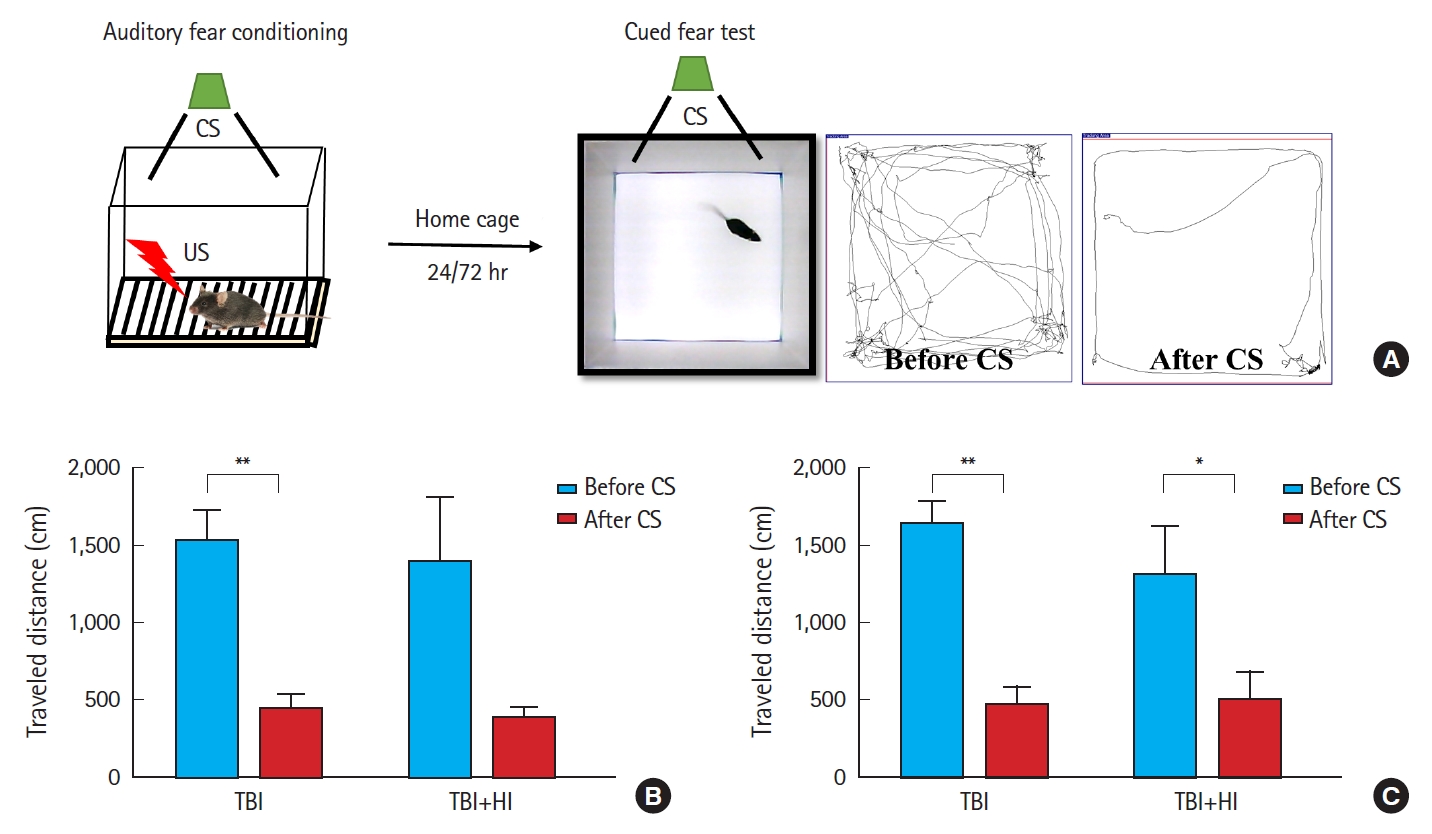
Acquisition of cued fear was assessed as locomotor activity changes due to freezing during tone presentation following foot shock (Fig. 5A). A significant difference was observed in mean changes in locomotion during pre-cue and cue presentation between TBI-only and TBI+HI groups on days 1 (Fig. 5B) and 3 (Fig. 5C) after fear conditioning. TBI-only mice demonstrated good learning ability, as they avoided entering the chamber in which they received a foot shock on days 1 and 3. No significant changes in locomotion were observed in TBI+HI mice during pre-cue and cue presentation on day 1, indicating deficits in cued fear recall relative to TBI-only mice.
NORT
Potential changes in cognitive function caused by HI were assessed using the NORT (Fig. 6A). TBI+HI mice displayed significantly lower preference for the object moved to a novel place compared to the object that remained in the same (familiar) place; these mice spent 9.5% of object exploration time in the test phase with the object that was moved to a novel place (Fig. 6B). No significant difference in exploration time was observed in TBI-only mice between familiar and novel objects (Fig. 6B). Comparison of the novel object discrimination index percentage between TBI-only and TBI+HI mice revealed a trend towards impaired cognitive ability in TBI+HI mice (Fig. 6C).
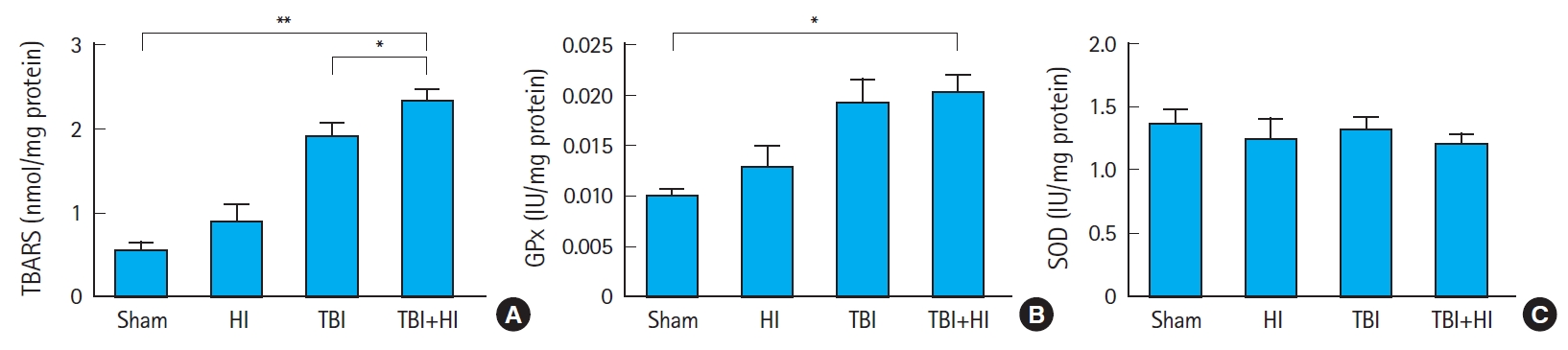
Oxidative stress following TBI
We measured the lipid peroxidation marker MDA (Fig. 7A), GSH levels as an indicator for glutathione peroxidase (GPx) activity (Fig. 7B), and SOD activity (Fig. 7C) as markers of oxidative stress, and compared the levels of these markers among groups 72 hours following surgery. MDA levels were significantly lower in TBI+HI mice than in TBI-only mice (Fig. 7A). GPx activity was significantly higher in TBI+HI mice than in sham counterparts (P<0.05) (Fig. 7B). No significant differences in tissue SOD levels were observed among groups (Fig. 7C).
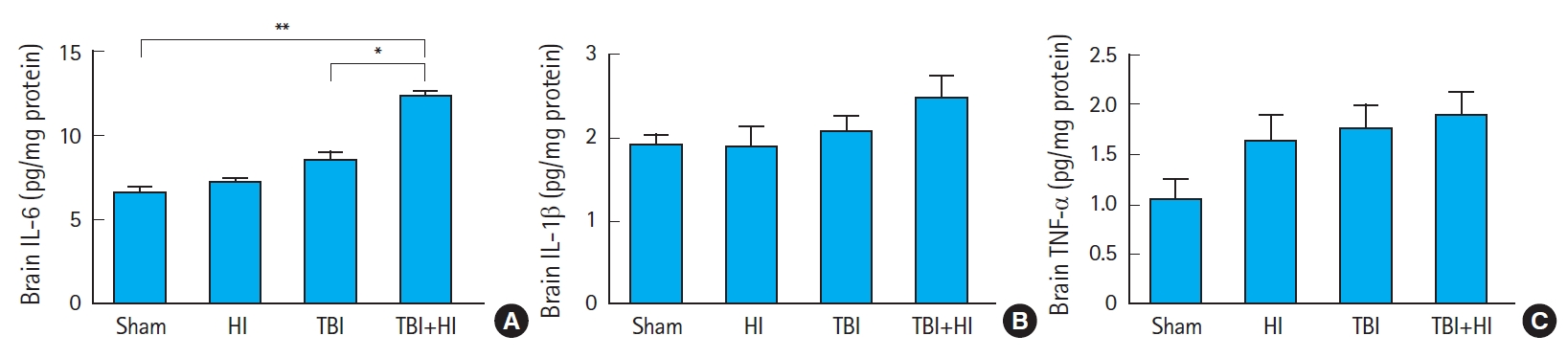
Proinflammatory cytokine expression after TBI
We compared the concentrations (pg/mg protein) of cytokines IL-6 (Fig. 8A), IL-1β (Fig. 8B), and TNF-α (Fig. 8C) among groups 72 hours postsurgery. IL-6 levels were significantly higher in TBI+HI mice compared to TBI-only and sham mice. We found no significant differences in tissue IL-1β and TNF-α levels between groups (Fig. 8B, C).
DISCUSSION
HI can inflict severe neurologic disability across all age groups. Birth asphyxia is the leading cause of neonatal encephalopathy, and cardiac arrest is the main cause of HI-mediated brain injury with subsequent morbidity and mortality [22,23]. The pathophysiology of this injury encompasses a heterogeneous cascade induced by the immediate cessation of cerebral blood flow [24], resulting in cellular energy failure, excitotoxicity, generation of free radicals, and apoptosis [22,25].
Systemic hypoxia following TBI simulates clinical situations of purely hypoxic insults such as airway obstruction, asphyxiation, and carbon monoxide poisoning, which exacerbate neurological deficits and increase lesion size, brain edema, and neuronal death [26-28]. However, the effects of post-traumatic HI after focal brain injury have not been addressed. We thus explored the impact of HI using a model of focal CCI followed by carotid occlusion alongside 60-min hypoxic ventilation.
Our results demonstrate that mice exposed to CCI and HI had a higher mortality rate and reduction in body weight than CCI-only mice. In addition, compared to CCI alone, post-traumatic HI increased neurobehavioral deficits in the BMT, CCFC, and NORT. These data resemble clinical findings in TBI patients, indicating that post-traumatic HI worsens neurological outcomes and prolongs recovery [5]. Our findings also highlight that a post-traumatic hypoxic insult has persistent detrimental effects on inflammatory responses and oxidative stress.
Cognitive deficits resulting from TBI include impaired memory and learning, attention, concentration, processing speed, executive functioning, and word-finding difficulties [29]. Factors contributing to TBI-induced memory impairments include alterations in neural circuits associated with memory function [30]. We previously reported that 2.0-mm CCI injury resulted in significant spatial learning and memory deficits when compared to sham animals [31]. Here, we employed three behavioral paradigms to assess learning and memory over 3 weeks. The BMT is an efficient cognitive task test to assess spatial/non-spatial learning following CCI injury in adult mice [31]. Compared to CCI alone, additional HI aggravated spatial learning and memory. Hippocampal lesions have deleterious effects on the ability to discriminate fear-associated contexts from a neutral environment [32]. A recent study reported that CCI does not affect the neural mechanisms of fear and extinction within the first 2 weeks of injury [33]. Using CCFC, we here demonstrate that compared to CCI alone, additional HI impaired fear-associated learning and memory. Unlike TBI-only animals, TBI+HI mice displayed significantly lower preference for the object moved to a novel place compared to the familiar one in the NORT, suggesting that HI affects memory function [34]. It is known that recognition memory is impaired in humans and animals with TBI [35,36]. Our results provide evidence that an additional secondary insult, such as HI, may further exacerbate TBI-inflicted damage.
HI injury to the mammalian brain can induce neurodegenerative responses. Unilateral HI mice exhibit unilateral hippocampal, cortical, and striatal tissue loss, and decreased density of hippocampal CA1 neurons. The hippocampus is susceptible to HI injury, and loss of hippocampal neurons may underlie HI-associated learning and memory deficits [37,38]. In our study, HI-only and sham mice showed comparable performance in the BMT, suggesting intact hippocampal function. This implies that HI injury through onevessel occlusion combined with 1-hour hypoxia in adult mice is sublethal. In contrast, we observed that HI induced significant learning and memory deficits following TBI.
Neuroinflammation is a prominent short-and long-term consequence of neuronal injury after TBI. It involves the activation of microglia and astrocytes, which release inflammatory mediators within the brain, and the subsequent recruitment of peripheral immune cells [39]. Animal models of TBI have been developed and proven valuable to elucidate the pathophysiology of the injury and enable preclinical assessment of the safety and efficacy of novel therapies [39]. Based on such models, it was shown that TBI induces a robust elevation of cytokines in the brain [40]. Our data did not reveal significant changes in brain cytokine production in TBI-only mice compared to sham controls 3 days after injury. In contrast, whilst IL-1β and TNF-α production remained unchanged, we observed a significant increase in IL-6 levels within TBI+HI brains compared to TBI-only and sham counterparts. IL-6 is involved in various physiological and pathophysiological processes, such as regulation of inflammation, immunity, bone metabolism, hematopoiesis, and neural development. IL-6 mRNA is upregulated within minutes of TBI, and increased IL-6 protein levels are detectable within an hour [40]. In this study, IL-6 levels were not different in TBI-only mice compared to sham mice, but were significantly increased in TBI+HI animals compared to TBI-only and sham counterparts 3 days following the injury. These results suggest that additional HI prolonged neuroinflammation, which may have already been cleared in TBI-only animals.
The intracellular antioxidant defense mechanism increases as an adaptive response to oxidative stress. Upon TBI, various oxidative stress markers such as lipid peroxides are produced in the brain, while antioxidant defense enzymes such as GPx and SOD are decreased in their activities; this imbalance is directly related to TBI pathogenesis [41]. GSH is a major thiol tripeptide that plays a crucial role in the cellular antioxidant defense system by scavenging free radicals and other ROS, removing hydrogen and lipid peroxides, and preventing oxidation of biomolecules [40]; we here measured GSH levels as an indicator for GPx activity. In the adult murine brain after TBI, GPx activity is increased within 24 hours after trauma [42]. In this study, we did not observe an increase in GPx levels 72 hours after TBI. However, when TBI was followed by HI, the activity of this enzyme was significantly elevated compared to sham mice, suggesting that HI exacerbated oxidative stress. Further studies are needed to examine time-dependent changes in oxidative stress markers. SOD is a crucial enzyme in cellular antioxidant systems that plays critical roles in the elimination of excess ROS in living organisms. It converts superoxide radicals to hydrogen peroxide, which is subsequently converted to water by catalase and GPx. In contrast to the change observed in GPx activity, SOD remained unchanged following TBI/HI.
MDA is an end-product formed during lipid peroxidation. A rise in its levels reflects an imbalance between endogenous antioxidants and oxidative stress. Elevated levels of MDA can be found in patients with TBI compared to controls, and MDA concentrations correlate with mortality following severe TBI [43]. In this study, we observed a significant increase in this lipid peroxidation marker in TBI+HI mice compared to sham and TBI-only animals 72 hours post-injury. Taken together, our findings suggest that posttraumatic HI significantly aggravated oxidative stress.
Overall, our study provides potential mechanisms underlying neurobehavioral outcomes following secondary HI insult after TBI. Thus, the prolonged neuroinflammation and oxidative stress in hippocampal neurons caused by HI resulted in aggravated spatial, fear, and discrimination memory deficits.
This study has several limitations. First, we used the one-vessel hypoxia model for HI, which does not take into account cardiac arrest. Second, we only impacted the focal right parietal lobe; further investigation of other types of focal brain and diffuse brain injuries is necessary. Finally, we did not administer impacts beyond 2.0 mm (2.5 or 3.0 mm) to avoid high mortality rates after surgery.
In summary, we provide evidence that secondary HI insult augmented spatial, fear, and discrimination memory deficits in an experimental TBI model. Results from this study suggest that TBI+HI-mediated neurobehavioral deficits may be at least partly explained by increased neuroinflammation and oxidative stress. Understanding these effects is an important first step in elucidating HI-associated pathophysiological progression and the foundation for developing appropriate therapeutics for post-traumatic HI.